





Pulmonary hypertension (PH) is said to occur when the mean pulmonary arterial pressure (mPAP) exceeds 25 mmHg at rest or 30 mmHg during exercise. There are many diverse causes of this condition but the term pulmonary arterial hypertension (PAH) is used to describe a rare group of diseases that share histopathological similarities in the small muscularised pulmonary arterioles leading to vascular remodelling known as plexogenic pulmonary arteriopathy (PPA). As a consequence, there is a progressive elevation in the pulmonary vascular resistance (PVR) that, if untreated, leads to death as a consequence of progressive right heart failure. Currently, there is no effective cure for PAH and the majority of treatments available either stabilise the condition or slow the rate of progression.1,2
Unfortunately, PH is often not considered or misdiagnosed and a median time of 14 months from symptom onset to diagnosis has been reported.3
It is essential that there is an improvement in the awareness of PH among health care professionals and that clinicians managing these complex patients know how to correctly interpret the investigations necessary to reach the diagnosis. Furthermore once the diagnosis has been made, patients should be referred to a specialist PH centre to enable appropriate therapeutic intervention to be made as soon as is possible.4
Classification
Although many conditions are associated with the development of PH, the PAH group is associated with the greatest elevation in PAP. Over the past 15 years a number of efforts have been made to classify conditions into groups according to common pathological and clinical features.5,6 These are outlined in Table 1.6
The PAH disorders are in group 1. At the present time, specific targeted therapy for PH is reserved for patients in groups 1 and 4. Group 1 patients share the histopathological entity known as PPA.7 In this condition, smooth muscle cells from the inner aspect of the media of muscularised pulmonary arterioles migrate though the intima into the lumen and proliferate. Once inside the lumen they differentiate into myofibroblasts, which are capable of laying down either fibrous tissue or smooth muscle tissue. The vascular proliferation develops in a concentric fashion so that on section the vessel has the appearance of an onion. Therefore, this type of proliferation is often referred to as onion skin proliferation. As the radius of the vessel gets less, the flow is reduced in proportion to the fourth power of the radius in accordance with Poiseuile’s Law. The pressure progressively rises and at points of proximal weakness where blood vessels branch, the wall gets progressively thinner and ultimately ruptures. Primitive blood vessels then grow into this area in a haphazard (plexiform) fashion giving rise to a plexiform lesion. The combination of concentric laminar (or onion skin) proliferation and plexiform lesions is known as PPA. Why this only occurs in those disorders listed in group 1 is far from clear although there are many theories relating to genetic predisposition, infection, autoimmunity and the role of pulmonary endocrine cells.1,4,7

Pulmonary thromboembolic disease can have many potential underlying causes including malignancy and thrombophillia abnormalities. Chronic thrombolembolic PH (CTEPH) is also an indication for advanced pulmonary vaso dilator therapy and for selected patients surgery in the form of pulmonary thromboendarterectomy may be an option.4
The pulmonary vasculature is normally a low-pressure low-resistance system with high distensibility as the resistance vessels normally possess only a small amount of smooth muscle relative to their systemic counterparts.7 This enables the right ventricle to deliver the same stroke volume as the left ventricle with 1/6 of the stroke work.8 However, as vascular remodelling occurs and vascular obstruction develops the resistance to flow increases.
The PVR is calculated by the following equation:
PVR = mPAP – mean pulmonary capillary wedge pressure (mPCWP) Cardiac output (CO)
As the PVR progressively increases in patients with PH, the right ventricle is susceptible to pressure overload and the response of the right ventricle to this increased after load determines the patient’s exercise capacity, symptoms and outcome.9,10 Ultimately patients die from right ventricular failure.
Over 90 % of patients do not fall within group 1 and left heart disease is one of the most common causes of PH in general.11,12
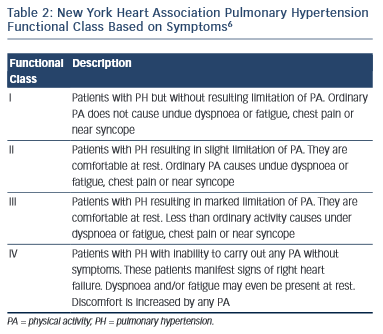
The functional class of patients can be determined using the New York Heart Association (NYHA) criteria and this can be used to help determine disease severity and prognosis.
Symptomatology
The diagnosis of PH is frequently missed. When it is associated with other comorbidities e.g. lung or heart diseases failure of the primary disease to respond to conventional therapies should lead one to suspect a possible association with PH in addition which warrants diagnosis, classification and potential therapeutic intervention.
For many patients symptoms develop late in the course of the disease and they are often non-specific. As a consequence, delay or failure of diagnosis is still far too common. Symptoms that are associated with PH include dyspnoea with exertion, which may progress and subsequently occur with minimal exertion, or may occur at rest as the PVR rises. Palpitations are often due to right atrial stretch and underlying atrial arrhythmias. Haemoptysis may reflect vessel rupture as elevation in PAP progresses or indeed can be associated with pulmonary thromboembolic disease. Pre-syncope and syncope are common as the PVR progressively rises. Certain conditions including hypoxia or general anaesthesia can lead to an acute elevation in PVR over a short period of time resulting in compromised left heart filling with low cardiac output and profound hypotension or even death. This is termed a PH crisis. Chest pain is often attributed to right ventricular angina as the right ventricle hypertrophies.
In addition to the NYHA classification of dyspnoea, an unencouraged 6-minute walk test is a useful benchmark and can be used to monitor a patient’s progression.
History and Examination
It is important to consider the diagnosis of PH and to take a careful history e.g. a familial background of PH, history of connective tissue disease or of thrombophilia abnormalities, alcohol consumption, etc. Physical examination may reveal evidence of other pathologies e.g. left heart failure, underlying lung disease, deep venous thrombosis, connective tissue disease or malignancy.
The clinical signs associated with PH include:
1.A prominent a wave reflecting high right ventricular filling pressures in the jugular venous pressure.
2.An accentuated V wave suggesting tricuspid regurgitation in the jugular venous pressure.
3.A loud pulmonary component of the second heart sound as a consequence of forceful valve closure from raised PAP.
4.A left parasternal lift as a consequence of right ventricular hypertrophy (a right ventricular heave).
5.A right ventricular gallop rhythm (third and or forth heart sound).
6.Signs of right ventricular failure.
Investigations
Blood Tests
Routine haematological and biochemical parameters are performed including autoimmune profile, HIV serology and thrombophilia screen (if pulmonary embolism is suspected). Genetic studies are important if familial PAH is being considered. B-type natriuretic peptide (BNP) or N-terminal pro b-type natriuretic peptide (NT-proBNP) can be elevated in PH and mainly in the absence of left heart disease may suggest the diagnosis or be used to monitor right ventricular dysfunction.1,2,4,13 Both are unhelpful in the diagnosis of early disease before there is significant cardiac dysfunction. BNP also rises in response to left ventricular failure and so it is not specific for the right heart.
Chest Radiology
Chest radiography has low sensitivity and specificity for the evaluation of PH, but does provide a useful, rapid and repeatable overview of cardio-pulmonary status. It may of course suggest that a lung pathology is causative.1
Electrocardiogram
Right atrial and right ventricular overload may be identified on a twelve-lead electrocardiogram (ECG), P wave amplitude can be assessed and abnormal QRS features may be noted however a normal resting ECG does not exclude PH. Arrhythmias may also be present particularly if there is right atrial stretch and 24-hour Holter monitor may be indicated if confirmation of underlying cardiac arrhythmia is necessary.1,4
Echocardiography
Echocardiography is a useful investigation in the initial diagnosis of PH and helps to risk stratify patients and monitor progress. It is also helpful in assessing right and left heart interaction and other potential causes of PH such as intracardiac shunting, valvular heart disease or primary myocardial disease.1,4,14 The commonest method of assessing pulmonary artery pressure is the use of Doppler echocardiography. This determines right ventricular systolic pressure, which is equivalent to pulmonary artery systolic pressure if there is no obstruction to the right ventricular outflow tract. The right ventricular systolic pressure is calculated by measuring the maximum tricuspid regurgitant jet velocity and then the modified Bernoulli equation is applied to give the trans-tricuspid valve pressure gradient. The right atrial pressure is added to the trans- tricuspid valve pressure gradient to calculate right ventricular systolic pressure. However an estimate of pulmonary artery systolic pressure should always be interpreted alongside an assessment of right ventricular size and function (e.g. using the tricuspid annular plane systolic excursion [TAPSE]) and within the clinical context. Even in experienced hands echocardiography can be unreliable in diagnosing or excluding PH and often the derived pressure measurements do not correlate well with measurements subsequently obtained at right heart catheterisation. Furthermore, it is possible for patients to have PH in the absence of significant tricuspid regurgitation, which can render assessment of pulmonary haemodynamics difficult. Therefore, current guidelines support determining only a probability of PH from echocardiographic evaluation. This study however does play a central role in disease surveillance evaluating how the right ventricle adapts over time and also enabling us to monitor response to treatment.
Lung Function Tests and Arterial Blood Gas Analysis
Pulmonary function testing is useful in diagnosing and assessing the contribution of underlying lung disease in patients with PH. In PAH the carbon monoxide diffusion as a percentage of predicted (diffusing capacity for carbon monoxide [DLCO] percentage) is usually impaired, independent of lung disease.1,4,6 It is important to bear in mind that patients who have unexplained dyspnoea with a low carbon monoxide diffusion capacity for which there is no obvious cause may have underlying pulmonary thromboembolism. Overall, there is no convincing correlation between lung function tests and pulmonary haemodynamics and therefore pulmonary function tests are not a reliable screen for patients with PH. They are of course important for patients who have primary lung diseases with PH resulting as a secondary phenomenon. There is some evidence that patients with scleroderma who have a reduced diffusion capacity for carbon monoxide are more likely to develop PH.15 Arterial blood gas analysis may be normal in patients with PH particularly those with PAH.
The Unencouraged 6-minute Walk Test
The unencouraged 6-minute walk test is a reproducible measure of peak oxygen consumption, is easily performed, is inexpensive and results correlate well with functional status and are an independent prognostic marker.16,17 A value below 300 metres may suggest a worse prognosis and mandate a review to change or initiate treatment,13 although it is not a reliable marker of pulmonary vascular disease progression.18 Cardiopulmonary exercise testing may also be used to functionally evaluate patients with PH and may also assist with prognosis and response to treatment.19,20
Other Radiological Imaging
Other radiological imaging includes ventilation perfusion (VQ) scanning, which is usually applied to investigate patients with PH potentially associated with pulmonary thromboembolism,21,22 but abnormalities are not usually helpful in confirming the diagnosis of PAH.21 However, the historical importance of VQ studies in distinguishing CTEPH from non-CTEPH causes of PH is diminishing with the use of multi-detector computed tomography (CT) (MDCT) scanning1,23 and in its greater resolution and disease-discriminating capacity. In terms of CT scanning, VQ imaging retains a radiation dose advantage and is a pure functional technique. Catheter pulmonary angiography was once considered the gold standard for the determination or exclusion of CTEPH in PH,24 but this is rarely performed now. It is usually limited to occasional cases where further delineation of complex congenital anatomy is required or where there is a concomitant need to perform an image-guided interventional procedure, such as mechanical clot disruption or biopsy of an indeterminate and potentially neoplastic intravascular lesion.1
Computed tomographic pulmonary angiography (CTPA) is the predominant current radiological imaging strategy for the evaluation of patients with suspected PH.25,26 MDCTPA provides the best temporal and spatial resolution providing a high-resolution comprehensive vascular and pulmonary evaluation in less than 5 seconds. Magnetic resonance imaging (MRI) imaging is complex, has limited availability and is associated with poor pulmonary parenchymal evaluation. Therefore, in the near future, this is not likely to replace CT evaluation.1
Right Heart Catheterisation
The gold standard investigation for diagnosing PH is right heart catheterisation.27–29 The procedure is usually performed via the right internal jugular vein although the femoral or subclavian veins are suitable alternatives. A balloon-tipped, triple-lumen Swan-Ganz catheter is connected to a pressure transducer and inserted through the venous sheath. Pressure measurements are then taken in the right atrium, right ventricle and pulmonary artery and the catheter is then advanced carefully with the balloon inflated and wedged in a more distal aspect of the pulmonary artery and the pulmonary capillary wedge pressure is measured. The balloon is then deflated and cardiac output measurements are made using the Fick method or more commonly an indicator dilution method.1 At least three readings of cardiac output should be obtained and the mean value is used to ensure accuracy. It is also routine to measure pulmonary artery oxygen saturation (Sv02) and when intracardiac shunting is suspected oxygen saturations are also measured in the right ventricle, at different points in the right atrium and in the vena cavae. The PVR is calculated as described above and the normal value is less than 2 Wood units (1 Wood unit is 1 mmHg per litre per minute and equates to 80 dynes. second.CM−5). Assessment of PVR is more helpful than mPAP alone when documenting the severity and pathophysiology of PH. As the disease progresses the cardiac output through the lungs falls and this is reflected in a lower pulmonary artery pressure value than is predicted by the patient’s clinical presentation. However when the PVR is calculated it may be demonstrated that the unexpectedly low pulmonary artery pressure is falsely misleading. This has important implications if surgical intervention is being considered for other co-morbidities in these patients.4
Cardiac index (litres per minute per body surface area squared) or PVR index can be calculated by adjusting for body surface. The body surface area is approximated from formulae, such as the Dubois formula, which incorporates height and weight.30 Vaso reactivity testing can be performed during right heart catheter to demonstrate a reduction in mPAP or PVR with an improvement in cardiac output. For some patients with PAH (usually less than 10 %) vaso reactivity testing may indicate a likelihood to respond to long-term calcium channel blockers and these patients may have a better prognosis.31 Those who have a positive response will demonstrate reversibility in mPAP by greater than 10 mmHg to achieve an absolute value of less than 40 mmHg with an unchanged or increased cardiac output during right heart catheterisation when they are given nitric oxide, prostacyclin or adenosine.1 Vaso reactivity testing is not reliable to predict response to treatment for patients who are classified in the other PH groups. It should only be performed by experienced operators cautiously with graded dosing in medically stabilised patients. It should not be performed in patients suspected of having pulmonary veno-occlusive disease as life-threatening pulmonary oedema may be precipitated.13
Most right heart catheter procedures are performed quickly (less than 30 minutes), are well tolerated and have a low complication rate.27 In our unit we also have a special protocol adopted to perform right heart catheter in patients with sickle cell disease.1
Pathophysiology
Our understanding of the molecular biology of the pathobiology of PAH has improved in recent years and this in turn has helped to develop new therapeutic agents against a variety of potential molecular targets. It is possible that vascular injury can occur in patients with PAH who have a genetic pre-disposition e.g. those with bone morphogenetic protein receptor 2 (BMPR2) mutations. Should these mutations occur there may be a loss of the inhibitory (regulatory) action of BMP on vascular smooth muscle growth. Should a subsequent insult occur e.g. as a consequence of autoimmune disease, drugs, HIV infection or toxins (which are not metabolised in patients with liver disease) vascular injury can occur. This may lead to endothelial cell dysfunction with, for example, abnormal production of nitric oxide, prostaglandins, endothelin 1, etc. Smooth muscle cell dysfunction and migration and proliferation due to abnormalities in the production of calcitonin and gastrin-releasing peptide or 5 hydroxytryptamine can follow,5 and lead to inflammation facilitated by a variety of interleukin, chemokines, fract alkaline and many others, which ultimately give rise to vascular remodelling and PPA.4
Disease Progression
As PH progresses the PVR rises and cardiac output falls. Initially dyspnoea may occur with exertion and subsequently at rest. Chest pain, palpitations, pre syncope, syncope or haemoptysis may be reported and ultimately the signs and symptoms of right heart failure develop. A median survival of 2.8 years has been reported for untreated patients in NYHA class 3 or 4.3,5,6 PH crises are acute elevations in PVR and are potentially life threatening as a consequence of acute reduction in left heart filling with profound systemic hypotension. This can occur during general anaesthetic induction (e.g. when the systolic vascular resistance falls) and is one of the reasons that patients with PAH need careful preoperative evaluation prior to surgical intervention.
Surgical Intervention
Pre-operative close communication between the PH team, surgeons and anaesthetists is essential when patients with PH require surgical intervention. The patient may require deployment of a PA flotation catheter perioperatively, advice may be required on fluid replacement, optimal haemoglobin values inotropic and vasoactive support and how baseline therapy will be given if patients are intolerant of oral drugs post op. It is important that the patient is managed in an appropriate environment e.g. for some patients post-operative management in intensive care may be appropriate and that appropriate work up and treatment strategies are defined. A joint discussion regarding the most appropriate approach to surgical intervention e.g. open versus laproscopic should take place. For example, laparoscopic abdominal surgery may require intra-abdominal gas inflation and this may compromise venous return, which in turn may be poorly tolerated. Additionally, pre-operative close communication and assessment will ensure that if a pulmonary hypertensive crisis develops, appropriate information will have been given to staff who will have management strategies in place. Whenever possible, patients with congenital heart disease should be managed in a congenital heart disease centre. Pregnant women with PAH should always be supervised closely by a PH centre and delivery should ideally be made at a specialist centre or have a PH specialist as part of the multi-disciplinary team in attendance.32
Depending on the underlying cause of PH and the kind of surgery proposed, mortality rates between 7 % and 24 % have been reported especially in cases of emergency interventions.32–35 It is clear that PH must be diagnosed, classified and treated pre-operatively and that there is pre-operative multi-disciplinary input. The necessity for surgery must be scrutinised bearing in mind the potential risks attendant on the proposed procedure for the patient and their median-term outcome on the basis of their PH alone independent of intervention. Pre-operative optimisation includes identifying and addressing conditions contributing to PH and assessing whether peri- and post- operative pulmonary vaso dilators are necessary. It is important that PH specialists are involved in the pre-operative treatment strategy.
Treatment
If patients have PH occurring in association with other disease processes their primary disorder should be optimised in the first instance. Strategies for the treatment of PAH vary around the world and typically advanced pulmonary vasodilator therapy is given to those patients who are in groups 1 and 4 and those who are in renal failure on dialysis. It is customary for advanced pulmonary vaso dilator therapy to be prescribed if the mPAP exceeds 25 mmHg at rest and the mean pulmonary capillary wedge pressure is less than 15 mmHg. Usually patients who receive specific targeted therapy are in World Health Organization (WHO) functional class 2, 3 or 4.1,2,4,36
There is insufficient evidence to justify the use of specific targeted therapy to treat patients whose PH occurs in association with lung disease. Furthermore, there is concern that should patients have PH in association with cardiac disease with left atrial filling pressure above 15 mmHg, specific targeted therapy may lead to increased venous return that, in turn, can exacerbate or produce acute left heart failure. Patients with obstructive sleep apnoea should have appropriate treatment e.g. lifestyle advice, nocturnal nasal continuous positive airway pressure in the first instance and only have consideration given to advanced pulmonary vasodilator therapy should PH persist after standard therapies for sleep-disordered breathing have been prescribed.1,4
For the majority of patients who develop PAH there is no effective cure, but the use of specific targeted therapeutic agents have been shown to improve exercise capacity, WHO functional class, haemodynamic parameters and time-to-clinical worsening. Recent evidence suggests that one of the newer endothilin receptor antagonists (ERA), macitentan, may be associated with improved survival, although further studies are required to support this.37 Usually ERAs are prescribed as second-line therapy after patients have been started on either a calcium channel blocker or much more commonly a phosphodiesterase type 5 inhibitor. ERAs are contraindicated in pregnancy because of concerns relating to teratogenicity.
A small number of PAH patients may respond to calcium channel blockers; however, calcium channel blockers are not effective for the vast majority of patients with PAH. They should only be prescribed either by or in conjunction with PH specialists and should not be given as an alternative to recommended therapies.
Sildenafil and tadalifil are phosphodiesterase type V inhibitors acting on the nitric oxide pathway promoting vaso dilatation. Sildenafil may also possess anti-proliferative effects on vascular smooth muscle.37–39 Soluble guanylate cyclise stimulators (e.g. riociguat) reduce intracellular calcium in a nitric oxide dependent and independent fashion and have been used to treat patients with PAH and those whose PH is associated with CTEPH.40
Usually patients are started on a phosphodiesterase V inhibitor and then either an ERA is added or replaces the initial agent. There are different ERAs available that can block either the endothelin A or A and B receptors and these antagonise vaso contriction and vascular remodelling promoted by the excessive endothelin release known to occur in patients with PAH.1,2,4,6,36–39
As a consequence of other endothelial factors that are absent or deficient in patients with PAH some patients, usually those in WHO class 3 who are unresponsive to combination therapy or in class 4 can be given intravenous continuous prostacyclin therapy, although this agent can also be prescribed by regular inhalation of subcutaneous injection.4 A newer oral formulation is expected to be available for clinical use within the next 2 years. However, the precise role of combination therapies for patients with PAH is receiving constant evaluation. There are a variety of other agents being studied, which include anti-inflammatory drugs, monoclonal antibodies, anti-platelet agents and lipid-lowering compounds.4,36
For some patients in class 4 disease, atrial septostomy is available to offload the right ventricle as a bridge to transplantation.40
Pulmonary thromboendarterctomy is available for selected patients with CTEPH although this is a major operation with a complication rate approximating 50 %. It should be performed in specialised centres.4,36 Lung transplantation (single or more usually bilateral) is an option for carefully selected patients, but limited donor organ availability and the complication of obliterative bronchiolitis remain major problems to be addressed.
Conclusion
It is hoped that further improvement of our understanding of the pathophysiological mechanisms involved in the development of PAH will be accompanied by the development of more effective treatments and that the role of combination therapy will be defined. However, PAH remains a progressive lethal disease that is frequently not diagnosed because initial symptoms are non-specific. Improved awareness is therefore necessary to ensure that patients with PAH receive earlier diagnosis and are referred to an appropriate specialist centre to have the condition characterised and to receive appropriate therapeutic intervention as soon as possible. It is widely held that patients with PAH should be managed in a PH centre where there are specialist clinicians and nurses trained in the assessment and management of these challenging patients. Often this model occurs in a combination or shared cared arrangement with one or more satellite hospitals who have agreed shared protocols, joint MDT meetings and clinics. This offers convenience for the patients and should they develop problems locally they can attend their local hospital and have appropriate advice and treatment given. If necessary, support can be sought from the primary specialist centre. Patients who are pregnant require careful MDT assessment and regular antenatal and perinatal input from the PH team. Likewise patients with PH who are referred for surgical intervention require close communication between anaesthetists, surgeons and the PH team.