





The development of heart failure is rarely dependent on primary alterations of cardiac metabolism. The majority of heart failure cases result from diseases of the cardiac muscle, most frequently ischaemic heart disease. However, whatever the cause of heart failure, the net result will be depletion of myocardial adenosine triphosphate (ATP), phosphocreatine and creatine kinase levels with decreased efficiency of mechanical work. Once heart failure has developed, the neurohormonal axis is activated with the aim to sustain haemodynamic failure. Activation of adrenergic and renin-angiotensin-aldosterone systems indirectly determine specific metabolic alterations in the cardiac and skeletal muscles. Over the last two decades, despite the adoption of drugs able to block neuro-hormonal activation in heart failure dramatically improving the overall prognosis of this deadly disease, mortality and morbidity remain a critical problem. In fact, apart from the well-known effects on chronotropism, inotropism, vascular tone and blood volume, the residual physiological effects of neuro-hormones indirectly determine a state of low metabolic efficiency in both the skeletal and cardiac muscles. The aim of this review is to analyse the metabolic derangement in the failing heart and, on this basis, speculate on possible new therapeutic targets.
Deranged Cellular Metabolism in Heart Failure
Under normal conditions, the healthy heart derives most of its energy from the free fatty acid (FFA) pathway that accounts for approximately two-thirds of energy production; the other source of energy being derived from glucose oxidation and lactate.1,2 FFA and glucose metabolism inter-regulate each other, a process referred to as the Randle cycle.3 Increasing FFA oxidation in the heart decreases glucose oxidation, while increasing glucose oxidation inhibits FFA oxidation. However, energy being derived from FFA oxidation is a less efficient source of energy than glucose oxidation (in terms of ATP produced per O2 molecules consumed) and determines a reduction of cardiac efficiency. In fact, the amount of ATP produced per O2 consumed is greater when glucose is oxidised compared with FFA and, therefore, FFA is a less efficient energy substrate than glucose. Elevated FFA oxidation can result in up to a 30 % decrease in cardiac efficiency.2
Progressive heart failure induces an imbalance between the requirement of cardiac tissue for oxygen and metabolic supplies and their availability, resulting in functional, metabolic and morphological alteration of the myocardium. At a cellular level, glucose uptake is decreased and conversion to lactate is increased; lactate uptake by the heart is switched to lactate production, and pyruvate is mostly transformed into lactate, thereby increasing cell acidosis. The FFA pathway is also slowed down, yet most of the produced energy comes from FFA oxidation, resulting in less ATP production. These metabolic changes lead to disruption of cell homeostasis, alterations in membrane structure and, ultimately, cell death. The following sections will attempt to clarify the mechanism at the base of these metabolic changes.
Effects of the Neuro-hormonal Activation on Metabolism of the Failing Heart
Neuro-hormonal activation significantly contributes to cardiac mechanical and metabolic inefficiency of the cardiac muscle and whole body of patients with heart failure. This vicious circle is likely mediated by increased use of non-carbohydrate substrates for energy production,2 resulting from different mechanisms. Adrenergically mediated increased peripheral lipolysis (wasting of subcutaneous fat and skeletal muscle) results in grossly augmented FFA availability. In fact, fasting blood ketone bodies4 as well as fat oxidation during exercise5 have been shown to be increased in patients with heart failure. Adrenergic activation may also induce insulin resistance, which is also associated with heart failure6 and may further contribute to increased circulating FFA levels by the development of ketosis and consequent impaired suppression of lipolysis. Indirect effects of augmented adrenergic tone in heart failure include increased heart rate, vasoconstriction and inotropism, which, in turn, may also indirectly contribute to a state of functional and metabolic inefficiency.
Angiotensin II is also an important regulator of cardiac energy metabolism and function.7 There are several mechanisms through which angiotensin II contributes to heart failure occurrence and persistence. Angiotensin II damages mitochondria in the cardiomyocyte by increasing reactive oxygen species production8 and affects mitochondrial oxidative phosphorylation, including FFA oxidation.9,10 These data suggest that angiotensin II affects FFA oxidation. There is also evidence that angiotensin II regulates glucose oxidation7,11 and its inhibition may exert beneficial effects. In addition, by decreasing oxidative metabolism, angiotensin II can compromise ATP production, thus reducing its availability.12 In this context, angiotensin II antagonism represents an attractive therapeutic approach. Studies using the euglycaemic insulin clamp technique have indicated that the beneficial effect of angiotensin II is exerted on insulin sensitivity. In fact, angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor antagonists have been shown to improve both left ventricular function and glucose homeostasis.13,14 Increased blood flow in skeletal muscle, accumulation of bradykinin or more efficient insulin release may be suggested as potential modes of action.
Endothelial dysfunction, a critical component in the progression of heart failure, may result from increased oxidative stress, secondary to activation of the adrenergic and the renin-angiotensin systems and to the production of inflammatory cytokines.15 In heart failure, the role of reduced bioavailability of nitric oxide (NO) is still under debate,16,17 while increased endothelin-1 (ET-1) levels are a mainstay.18 Growth factors, vasoactive substances and mechanical stress contribute to the increased ET-1 levels in patients with heart failure. Despite the known adaptative aspect of supporting contractility of the failing heart, persistent increases in cardiac ET-1 expression in the failing heart have a pathophysiological maladaptive aspect and are associated with the severity of myocardial dysfunction.19 It has been observed that trimetazidine could reduce endothelin release in patients with cardiac disease.20,21 Trimetazidine-induced reduction of intracellular acidosis in ischaemic myocardium might not only influence myocardial but also endothelial membranes.22 By decreasing endothelial damage, trimetazidine could inhibit ET-1 release that, in turn, may decrease myocardial damage. A second hypothesis is that, by just decreasing the effects of chronic myocardial ischaemia, trimetazidine could inhibit ET-1 release. Therefore, the observed decrease in ET-1 release with trimetazidine, could likely be linked to trimetazidine-induced reduction of myocardial ischaemia. Finally, keeping in mind the close relation between endothelium and insulin sensitivity, the observed effects of trimetazidine on endothelial function could also explain the beneficial action of trimetazidine on glucose metabolism.
In the same context, the potential beneficial effect of 6 weeks of oral L-arginine supplementation on endurance exercise, an important determinant of daily-life activity in patients with chronic stable heart failure, has been assessed.23 L-Arginine is the precursor of endogenous NO, which is a potent vasodilator acting via the intracellular second-messenger cyclic guanosine monophosphate. In healthy individuals, L-arginine induces peripheral vasodilation and inhibits platelet aggregation due to an increased NO production. The results of this study show that arginine enhanced endurance exercise tolerance, reducing both heart rate and circulating lactate levels, suggesting that chronic arginine administration might be useful as a therapeutic adjuvant to improve the patient’s physical fitness.
In summary, in the failing heart neuro-hormonal activation determines a combination of direct and indirect haemodynamic and metabolic actions, which, despite a potential teleological purpose, will eventually lead to further deterioration of cardiac function, mainly mediated by the resulting decreased metabolic efficiency of the cardiomyocytes. Specific therapies may attenuate these effects.
Abnormal Glucose Metabolism in Heart Failure
As glucose and lactate are more efficient fuels for aerobic respiration, increasing the use of these substrates can improve the oxygen consumption efficiency of the myocardium by 16–26 %.24 In addition, skeletal muscle glucose uptake in the heart and arm is inversely related to serum FFA levels25 and increased FFA flux from adipose tissue to non-adipose tissue amplifies metabolic derangements that are characteristic of the insulin resistance syndrome.26 Further findings suggest that raised FFA levels not only impair glucose uptake in heart and skeletal muscle but also cause alterations in the metabolism of vascular endothelium, leading to premature cardiovascular disease.27
Global Energy Expenditure in Heart Failure
Energy consumption at rest appears higher in patients with heart failure than in healthy subjects.28–30 It has been shown that increased rate of energy expenditure is related to increased serum FFA oxidation and that both energy expenditure and serum FFA oxidation are inversely correlated with left ventricular ejection fraction and positively correlated with growth hormone, epinephrine and norepinephrine concentrations.31 Norepinephrine increases whole-body oxygen consumption, circulating FFA concentrations, and FFA oxidation.32 These changes have been attributed to stimulation of hormone-sensitive lipase in adipose tissue, and to stimulation of oxygen consumption independent of lipolysis by norepinephrine.33 These data, together with close correlations between plasma norepinephrine concentrations, energy expenditure at rest and FFA oxidation, make increased sympathetic activity the most likely explanation for alterations in fuel homeostasis in patients with heart failure.33 Therefore, intervention strategies aimed at optimising global and cardiac metabolism could be useful for interrupting the vicious circle of reduced function at greater metabolic expenses in different cardiac conditions.
Pharmacological Implications of Impaired Myocardial Metabolism in Heart Failure
Given the above-described pathophysiological background and the difficulty of standard treatment to control the total symptomatic and prognostic burden in many patients with heart failure, it seems logical to consider pharmacological manipulation of cardiac energy metabolism as an adjunctive therapeutic option. Optimisation of cardiac energy metabolism is based on promoting cardiac glucose oxidation.
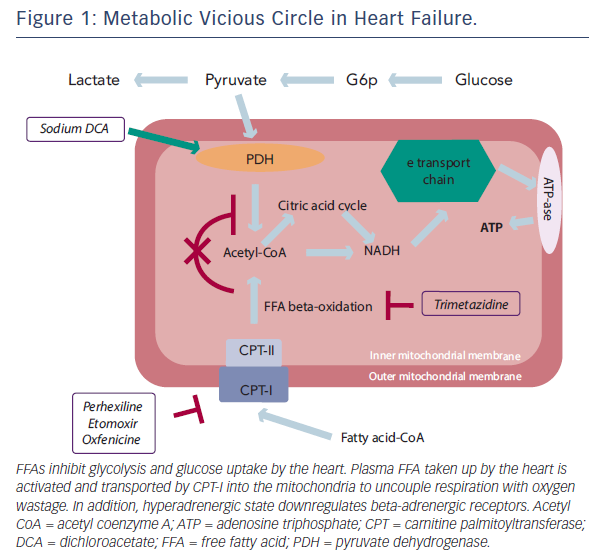
Stimulation of myocardial glucose oxidation can be achieved either directly with stimulation of glucose metabolism, or indirectly through inhibition of fatty acid beta-oxidation, in order to shift energy substrate utilisation away from fatty acid metabolism and towards glucose metabolism which, as explained above, is more efficient in terms of ATP production per mole of oxygen used. Therefore, metabolic therapy could play a beneficial role in terms of glucose metabolism homeostasis.
The concept that drugs able to promote the use of glucose and non-fatty substrates by the mitochondria may increase metabolic efficiency and function of the failing heart has prompted several clinical studies. Experimental studies have first shown that stimulation of pyruvate dehydrogenase activity leads to enhanced glycolysis and use of lactate by the myocardium for aerobic respiration.34 Myocardial consumption of FFA is simultaneously inhibited, with the overall effect of a change of substrate use from predominantly non-esterified FFA to glucose and lactate,35 finally resulting in improved left ventricular mechanical efficiency.36
Trimetazidine (1-[2,3,4-trimethoxybenzyl]piperazine dihydrochloride) has been shown to directly inhibit FFA oxidation by blocking 3-ketoacyl-coenzyme A thiolase (3-KAT), the last enzyme involved in beta-oxidation,37 although this issue remains controversial.38,39 Trimetazidine affects myocardial substrate use by inhibiting oxidative phosphorylation and by shifting energy production from FFA to glucose oxidation (see Figure 1).40 Several studies have outlined the potential benefits of this agent on regional and global myocardial dysfunction.41–49 3-KAT inhibitors could also play a beneficial role in terms of glucose metabolism homeostasis at both cardiac and skeletal muscle level. The beneficial effect of trimetazidine on left ventricular function, has been attributed to preservation of phosphocreatine (PCr) and ATP intracellular levels.50 Clinical studies using phosphorus-31 magnetic resonance spectroscopy to measure PCr:ATP ratios in human myocardium have shown that this ratio is reduced in failing human myocardium.51 The PCr:ATP ratio is a measure of myocardial energetics and its reduction may depend on imbalance of myocardial oxygen supply and demand,52 and reduction of the total creatine pool, a phenomenon known to occur in heart failure.53 In a study performed in patients with heart failure of different aetiologies receiving full standard medical therapy, it was observed that the trimetazidineinduced improvement of functional class and left ventricular function was associated with an improvement of PCr:ATP ratio, supporting the hypothesis that trimetazidine may preserve myocardial high-energy phosphate intracellular levels.54 These results appear particularly interesting, especially in view of previous evidence indicating the PCr:ATP ratio as a significant predictor of mortality.55 In fact, imetazidine has been shown to improve prognosis in patients with heart failure in a multicentre retrospective cohort study56 and in two meta-analyses.57,58 On this basis, its use in patients with heart failure has been advocated in a recently published position paper.59
Similarly to trimetazidine, ranolazine has also been shown to significantly improve left ventricular performance in experimental models of heart failure.60–63 Sabbah et al. measured haemodynamics before and 40 minutes after intravenous ranolazine administration in a canine model of heart failure.60 Results in 13 experimental dogs were compared with those obtained in eight normal healthy dogs. Ranolazine significantly decreased left ventricular end-diastolic pressure and increased left ventricular ejection fraction in the absence of any effects on heart rate or blood pressure. In subsequent experiments from the same laboratory, Chandler et al. reproduced these findings and determined that the improvement in left ventricular performance was not associated with an increase in myocardial oxygen consumption (MO2) compared with an intravenous infusion of dobutamine that improved left ventricular performance to a similar extent, but was associated with a significant increase in MO2 requirements.61
Overall, these data confirm that selective inhibition of 3-KAT represents a new therapeutic window in the treatment of patients with heart failure of different aetiologies.
Combined Metabolic Action of Beta-blockers and Trimetazidine
ACE inhibitors and beta-blockers remain the clinical mainstay of the treatment of heart failure. It is interesting to note that beta-blockers may yield an ancillary metabolic effect. Their principal mechanism of action is based on reduction of oxygen consumption by reduced heart rate and inotropism. However, a direct complementary metabolic effect could be exerted by beta-blockers themselves, by reducing peripheral lipolysis and determining reduction of FFA availability. There is indeed evidence that beta-blockade can reduce FFA use in favour of greater glucose use in patients with cardiac disease.64 This change in myocardial energetics could provide a potential mechanism for the decreased MO2 and improved energy efficiency seen with beta-adrenoreceptor blockade in the treatment of ischaemic heart disease and heart failure.65
The issue of whether non-selective, compared with selective betaadrenoreceptor blockers are more efficient in shifting total body substrate use from lipid to glucose oxidation66 remains controversial.67 Nevertheless, a better metabolic disposition of non-selective betablockers may contribute to improved survival rates observed with their use.68 In addition, central inhibition of sympathetic nervous activity with moxonidine has been associated with increased mortality rates in patients with chronic heart failure.69 In fact, despite a significant reduction of cathecolamine spillover and, consequently, heart rate, moxonidine has been shown to increase FFA use and increase MO2 consumption.70 This could be the reason for the failure of central sympathetic inhibition in preventing death in long-term studies in patients with chronic heart failure. It also indicates that the predominant mechanism of action of beta-blockers in cardiac syndromes is likely related to mechanisms of action other than simple heart rate reduction. In patients with heart failure the magnitude of heart rate reduction may therefore be a marker of improved functional response following beta-blockade administration, a consequent effect rather than a mechanism. Nonetheless, a clinical trial in which the cardiac ‘funny’ (If) channel inhibitor ivabradine (a pure heart rate-lowering agent) was added to beta-blockade (SHIFT [Systolic Heart Failure Treatment With the lf Inhibitor Ivabradine Trial]) clearly demonstrated that the greater the heart rate reduction the greater the reduction of hospitalisation events in patients with heart failure.71 Therefore, apart from the importance of heart rate lowering per se, a complementary synergistic metabolic action of beta-blockers and trimetazidine can be hypothesised: whereas the former reduce FFA availability, the latter decrease their cardiac use. Overall, this drug-induced metabolic shift could reduce FFA oxidation and increase the flux through pyruvate dehydrogenase with a consequent energy-sparing effect.54,72
Additional data also suggest that the metabolic effect of trimetazidine may also take place in other organs and tissues.72 In fact, apart from a reduction of whole-body energy demand, a trend for a reduction of whole-body lipid oxidation and of fasting plasma FFA concentration has also been observed.72 This general metabolic shift could reduce the overall metabolic requirements of the body, resulting in an attractive adaptation strategy in the context of coronary and myocardial insufficiencies. Interestingly, beta-blockers have also been shown to exert a direct effect on whole-body metabolism. In trained athletes, beta-adrenergic blockade abolishes the marked increase in plasma glucose levels during intense exercise as a result of enhanced peripheral glucose uptake, with no significant change in glucose production.73 These effects of adrenergic blockade on glucose kinetics could be mediated by direct effects or indirectly through changes in lipid substrates and/or counter-regulatory hormones.
Other Inhibitors of Fatty Acids Oxidation
Etomoxir, perhexiline and oxfenicine are carnitine palmitoyltransferase I (CPT-I) inhibitors. CPT-I is the key enzyme for mitochondrial FFA uptake; its inhibition, therefore, reduces FFA oxidation and their inhibitory effect on pyruvate dehydrogenase. As a consequence, glucose oxidation is increased.74,75 Etomoxir, initially developed as an antidiabetic agent, has been observed to improve left ventricular performance of pressure-overloaded rat heart.76 These effects have been considered due to a selective modification of gene expression of hypertrophic cardiomyocytes.77 Etomoxir has also been shown to increase phosphatase activation, have a direct effect on peroxisome proliferator-activated receptor-alpha and upregulate the expression of various enzymes involved in beta-oxidation.77 The first clinical trial employing etomoxir in patients with heart failure showed a significant clinical and cardiac function improvement.78 In experimental animal studies, etomoxir has also been shown to improve glucose metabolism.79 However, the use of etomoxir may be limited by the observations that it may cause cardiac hypertrophy80 and oxidative stress.81
Analogous to etomoxir, perhexiline and oxfenicine, originally classified as calcium antagonists, reduce cardiac use of long-chain fatty acids by inhibiting CPT-I.82–84 They were initially developed as antianginal agents.85,86 However, they have since been employed in patients with heart failure. In a previous study, metabolic modulation with perhexiline improved maximal oxygen consumption at the cardiopulmonary exercise test, left ventricular ejection fraction, symptoms, resting and peak stress myocardial function, and skeletal muscle energetics.87 More recently, and similarly to trimetazidine, perhexiline has been shown to improve cardiac energetics and symptom status with no evidence of altered cardiac substrate use, further supporting the hypothesis of energy deficiency in heart failure and further consideration of metabolic therapies in its management.88 Therefore, similarly to 3-KAT inhibitors, CPT-I inhibitors may represent a novel treatment in patients with heart failure with a good safety profile, provided that the dosage is adjusted according to plasma levels.
FFA Inhibition in Older Patients
Age-related changes of mitochondria impair the human host cells homeostasis and contribute to the development of most common ageing diseases. Older subjects without overt cardiac diseases are prone to develop heart failure with preserved ejection fraction. Risk factors do not fully account for the aged heart functional loss that might be underlined by a common pathogenic denominator (i.e. cell energy alteration at mitochondrial level in organs requiring high energy). In older men without overt cardiovascular disease, the presence of prepathologic conditions (pre-hypertension, reduced insulin sensitivity, impaired myocardial contractile reserve, inadequate vasodilation due to endothelial dysfunction, reduced cardiomyocytes renewal, systemic inflammation and raised coagulation capacity) are possibly related to reduced mitochondrial function and density. Several studies have indeed shown reduced mitochondrial content and function with ageing, leading to the theory that decreased mitochondrial content and increased uncoupling with age compromises the energy state of the cell.89 Indeed, altered beta-oxidation increases the reliance on long-chain fatty acids relative to glucose with subsequent decrease of cellular metabolic efficiency at any given level of tissue activity. On this basis, in older patients with coronary artery disease, partial fatty acid oxidation inhibition by trimetazidine added to standard optimal medical therapy has been shown to improve reverse remodelling of chronically dysfunctional myocardium90 and improve cardiac symptoms and quality of life.91 The observed improvement could be related to increased cellular energy reserve,54 which could be pivotal in a context of ageing-induced reduction of mitochondrial efficiency.
The Importance of a Correct Metabolic Substrate Availability
It remains questionable whether metabolic substrate availability rather than pharmacological shift from fatty acids to glucose oxidation may be appropriate in patients with long-lasting heart failure.92 In fact, the anti-lipolytic drug acipimox, which reduces substrate availability and impairs fatty acid oxidation, has been shown to worsen left ventricular function in patients with idiopathic dilated cardiomyopathy.93 In addition, when substrate availability is acutely modulated during exercise testing in patients with stable coronary artery disease and preserved left ventricular function using a high-carbohydrate meal versus a high-fat meal, a lower ischaemic threshold and greater ischaemia magnitude is observed following the high-carbohydrate meal.94 Reduced lipid uptake and disposal in the setting of heart failure may represent therefore a maladaptive response. A recent study evaluated the metabolic and functional effects of high- and low-serum FFA availability in the presence of normal fasting serum glucose and insulin concentrations.95 In patients with chronic heart failure, short-term reduction in serum FFA concentration, while serum glucose and insulin concentrations remained closed to the fasting levels, induced an impairment of left ventricular energy metabolism and left ventricular function at rest. A two-fold serum FFA increment in the same experimental conditions did not induce any detectable change.95 Although in previous studies pharmacological manipulation of the heart substrates preferences has been shown to exert beneficial effects, these data demonstrate that direct deprivation of energy substrates is detrimental for cardiac metabolism and function. The clinical fallout is that metabolic manipulation at the systemic level may be considered in order to optimise the treatment of these patients; we also believe that special cautions should be considered when iatrogenic and acute changes of substrates and regulatory hormones of glucose and FFA metabolism are to be performed in these extremely vulnerable patients.
Conclusion
All cardiac syndromes may induce and be maintained by modifications of cardiac metabolism. Heart failure may be dependent on and promote metabolic changes in part through neurohormonal activation determining an increased use of non-carbohydrate substrates for energy production. This metabolic adaptation yields a state of metabolic inefficiency. The net result is depletion of myocardial ATP, phosphocreatine and creatine kinase levels with decreased efficiency of mechanical work. Different therapeutic approaches have been developed to manipulate cardiac energy metabolism. However, the most effective approach consists in modifying substrate use by the failing heart and includes several pharmacological agents. These agents have been originally adopted to increase the ischaemic threshold in patients with effort angina. However, the results of current research support the concept that shifting the energy substrate preference away from fatty acid metabolism and glucose metabolism could be an effective adjunctive treatment in patients with heart failure. Nevertheless, the exact role of metabolic therapy in heart failure is yet to be established, and a large multicentre randomised trial is necessary.