





The concentration of plasma low-density lipoprotein cholesterol (LDL-C) is reduced, while that of total high-density lipoprotein cholesterol (HDL-C) is increased, following inhibition of cholesteryl ester transfer protein (CETP). This combined effect has made inhibition of CETP an attractive pharmacological approach for reducing the residual incidence of cardiovascular disease (CVD) remaining after optimal LDL-lowering therapy. However, since HDLs represent a complex dynamic heterogeneous family of particles varying in size, composition and function, the concept is not without difficulties. CVD has been shown to alter the particle complexity, in terms of both relative abundance of size/density and composition. The effects of raising the plasma concentration of HDL-C on a background of ‘disease modified’ are largely unknown. Randomised clinical trials to date have failed to provide evidence of benefit for this approach. The CETP inhibitor, dalcetrapib, which showed no harmful effect, also had no ability to reduce LDL-C and the study may be considered as providing strong evidence that HDL raising per se is not an effective approach in reducing CVD. The remaining members of the CETP inhibitor family, currently in Phase III studies, are able to reduce plasma LDL-C and may, thus, provide a beneficial effect on CVD, as long as it is not reduced through the adverse effects of raising plasma HDL-C, which may well be dysfunctional. Subsequent analysis of these large randomised controlled trials, due to be published in 2016/17, may allow us to further understand the effect of CVD on the complex metabolism of HDL-C.
The Birth of the HDL Hypothesis
It is a well-established fact that elevated plasma concentrations of low-density lipoprotein-cholesterol (LDL-C) is one of the most important risk factors for developing coronary artery disease (CAD), and, eventually, coronary heart disease (CHD)1 and other forms of atherosclerotic cardiovascular disease (CVD). Targeting LDL-C reduction has been effective in lowering CVD risk, through the use of HMG-CoA reductase inhibitors (statins) for both primary and secondary CVD prevention as the first-line therapy, resulting in reduced CV events and overall mortality2,3. However, despite reduction in LDL-C, 18–41 % with moderate doses of statins and 40–60 % with higher doses or more potent statins, there remains a significant residual cardiovascular risk. In the late 1970s the inverse relationship between plasma concentration of HDL-C and CVD risk was identified4 and subsequent prospective data from the Framingham Heart and ARIC studies further supported this idea5,6. It was proposed that CHD risk was inversely related to plasma concentration of HDL-C, owing to the ability of HDL particles to remove cholesterol from developing atherosclerotic lesions, and thus, the HDL hypothesis was conceived. Subsequent in vivo studies, in which plasma HDL-C was raised by infusion or transgenic expression of human apolipoprotein AI (ApoAI) in rabbit and mouse models of atherosclerosis demonstrated a potent atheroprotective effect7–9. With this wide array of potential benefits, raising plasma HDL-C was seen as a promising new drug target.
Raising Plasma Concentration of HDL-C
Although lifestyle changes, such as vigorous exercise, smoking cessation and weight loss have been shown to moderately increase plasma concentrations of HDLs10–13, individuals with low plasma HDL-C are more likely to respond to pharmacological treatment that increases HDL. Whilst statins, fibrates and some thiazolidenediones have been shown to modestly increase plasma concentrations of HDL-C14–16, nicotinic acid or niacins have been used to provide a more robust increase by 15–16 %17,18. Niacin increases plasma HDL-C by inhibiting the putative hepatocyte HDL-C catabolism receptor, preventing HDL-C catabolism, and thereby increasing the half-life of circulating HDL-C18. However, neither of the two large randomised controlled clinical trials, AIM-HIGH and HPS2-THRIVE, were able to demonstrate a difference in the primary end point despite favourable changes in HDL-C concentration17,18. Although these data may question the ability to provide a cardioprotective effect through elevating plasma HDL-C, nicotinic acid and niacins have dramatic side effects, such as flushing, which may well compromise any benefits observed.
CETP as a Novel Drug Target to Raise Plasma Concentration of HDLs.
Development of a novel target for elevating plasma HDL-C concentration was derived from analysis of the understanding of the biochemistry of HDL metabolism and particle dynamics. CETP is a hydrophobic glycoprotein which can catalyse the transfer of cholesteryl esters, generated by lecithin:cholesterol acyltransferase (LCAT), principally in larger -HDLs, to other lipoproteins in exchange for triglycerides (TGs), derived primarily from very low-density lipoproteins (VLDL) or chylomicrons19 (Figure 1). This process of lipid exchange has been shown to increase the relative abundance of small lipid-poor pre -HDL particles, which play a primary role in the acquisition of cholesterol from cell membranes via the ABCA1 transporter (reverse cholesterol transport)20. These data seeded the idea that modulation of HDL particle abundance could increase the ability to remove cholesterol from peripheral tissues, and was further supported by numerous in vivo studies where inhibition of CETP in animal models prevented cholesterol-induced atherosclerosis21–23, whilst CETP gene transfer in mice (a species lacking CETP activity) increased lesion formation21. Although these findings were encouraging, subsequent studies in other models did not reiterate the protective effect of CETP inhibition shown previously24–26. However, these data are difficult to compare and may reflect disparity in means by which CETP is modulated and in the mechanisms involved in generating atheromatous lesions in the specific models. In an extremely elegant set of experiments, Brousseau and colleagues were able to show, that raising HDL-C using the CETP-inhibitor drug, torcetrapib, did so through an effect on delaying catabolism27. Both torcetrapib and niacin increase HDL by delaying catabolism, which, in turn, increases the half-life of HDL-C, but neither of these methods have led to a beneficial clinical effect. As HDLs are heterogeneous, dynamic particles, it is likely that their structure and function will be significantly modified by prolonging the circulatory half-life and would depend largely on the effect of the clinical status of the patient.
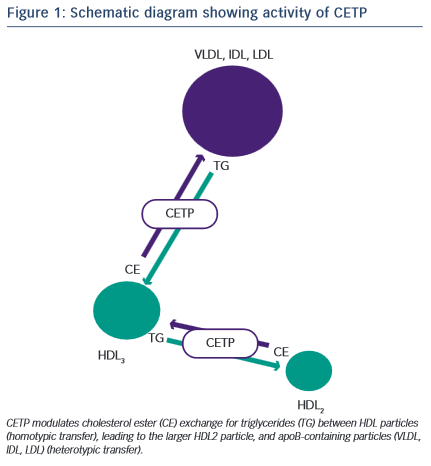
CETP Inhibition – Effect of Gene Polymorphisms
The human CETP gene has been mapped to chromosome 16 on the 16q21 locus, spanning about 25 kb and consisting of 16 exons and 15 introns. The current availability of DNA arrays capable of screening the entire genome for many thousands of SNPs in large cohorts has provided a new tool for studying the aetiology of complex diseases. However, the putative instrumental variables associated with alleles and clinical outcome are not always appropriate and the method is also limited by problems associated with linkage disequilibrium. Voight and colleagues reported a large Mendelian randomisation study confirming that lower CETP activity, when associated with both lower LDL-C and higher HDL-C, is associated with cardiovascular benefit28. Studies of CETP gene polymorphisms have been controversial. The B2 allele of the Taq1B SNP of the CETP gene (which is associated with low CETP activity and raised HDL-C) was found to be accompanied by a lower than average risk of CVD29. Further analysis showed that this association was limited to one ethnic group (Chinese)30. In a 10-year follow-up study of more than 18,000 women, among 350,000 SNPs, it was found that three SNPs in or close to the CETP gene (rs708272, rs432992 and rs7202364) were associated with an increase in plasma HDL-C and a reduced incidence of myocardial infarction in this population31. Current genetic analysis indicates that a decrease in CETP activity is correlated with a decrease in CVD risk.
Prospective Cohort Studies of CETP and CVD Risk
Measuring CETP activity is technically challenging and work by Ritsch and colleagues provides our sole reference data that there is a linear relationship between activity and concentration32. Prospective observational cohort studies to investigate the relationship between the plasma concentration of CETP activity or concentration and the risk of cardiovascular events generally show an inverse correlation, suggesting a negative association with CETP33–38. Of the three studies that evaluated populations free of CVD, two of them assayed CETP activity long-term, with a 10–15-year follow-up33,34, whilst the other study measured concentration and had a two-year follow-up35. The remaining three studies considered patients with clinical CAD or CHD and measured CETP activity36–38. Assessment of CETP activity or mass requires purification from total plasma or serum, which is then a measurement made in the absence of the proper biological complexity of whole blood and its many affecting factors; for example, the presence of ApoC-I, an isoform of apoliprotein C that has been recently demonstrated to be an endogenous inhibitor of CETP activity39. Although these prospective cohort studies are strongly suggestive that low concentrations/activities of CETP may correlate with increased CVD, these data are entirely reductive and do not to address the clinical effect of CETP inhibition in an appropriate patient population.
Currently, we have limited and insufficient precise knowledge of the complex dynamics between plasma lipoproteins and activation of vascular cells and cell membranes to explain the apparent paradox between the prospective observational epidemiology and genome- wide analysis for the role of CEPT inhibition. To enter into a discourse on whether the disparity is due to confounding effects or reverse causality would be fatuous at this point.
HDL Complexity and the Effect of Disease on HDLs
The potential for disease status to change the structure and, hence, function of HDLs is of paramount importance to our understanding of the subsequent effects of raising HDL-C concentration. Interpretation and prediction of the impact of CETP inhibition is complicated by the growing awareness that the effects of HDL-C may be modified in different clinical settings. Human plasma high-density lipoprotein (HDL) particles constitute a spectrum of pseudo-micellar protein/ lipid complexes, with hydrated densities within the range of 1.063 to 1.210 g/ml40. This spectrum may be further defined by five physicochemically-defined particle subpopulations determined by their buoyant density: HDL2b, 2a, 3a, 3b, and 3c. Compared with other lipoproteins, HDLs are protein-rich, with an average ratio protein:lipid of 1:1. Approximately 70 % of HDL protein mass is Apo AI, with Apo AII accounting for a further 15–20 %. Major components of the remaining 15–20 % of proteins are other amphipathic apolipoproteins (eg., ApoCI, ApoCIV, E, D, M and AIV), enzymes and lipid transfer proteins (eg paraoxonase (PON), PAF-acetlyhydrolase (PAFAH), LCAT, and CETP).
Proteomic analysis of HDLs, on the other hand, has been identifying putative mechanistic information for functional observations reported in previous decades. Recent proteomic studies have identified up to 49 proteins associated with centrifugally-isolated HDL41–44. Studies by Vaisar and colleagues44, investigating both total HDL and HDL3 from normocholesterolaemic control subjects and age-matched patients with CAD, identified a role for HDLs in protease inhibition in addition to an effect on complement activation, supporting findings from ten years earlier45. Data from a meticulous study, in which the proteome of HDL2b, 2a, 3a, 3b, and 3c, also from normocholesterolaemic subjects, were analysed, defined 28 distinct HDL-associated proteins which associated in clusters in subpopulations46. In this study the investigators were able to show that ApoL-I, PON1 and PON3 correlated with the capacity of HDL3 to protect against oxidation, confirming their results through measurement of the ability of HDL3 to reduce the rate of accumulation of conjugated dienes in an LDL oxidation assay. These observations confirm earlier functional studies in which PON was shown to be one of the major proteins responsible for the anti- oxidative function of HDLs47–49. In a recent report of a 10-year follow up of 88 type 2 diabetic patients, the incidence of cardiovascular events increased in proportion to reduced PON1 levels and activity, suggesting that PON1 may be an independent predictor of cardiovascular events in people with diabetes49. Modulation of the structure and function of HDL-C has been reported, initially with regard to the anti-oxidative function of the particle50, but also with respect to the anti-inflammatory, cholesterol transfer, and anti-thrombotic function of HDL-C51–53.
Clinical Outcomes, so Far
The first Phase III randomised controlled trial for a member of the CETP inhibitor family (Torcetrapib: ILLUMINATE; NCT00134264) began in July 2004 comparing 15,067 patients randomised to high-intensity statin alone or high-intensity statin plus torcetrapib54. This study was terminated early when it became clear that the drug increased the incidence of the primary CVD endpoint, despite raising HDL-C by 70 % and lowering LDL-C by 20 %. Following analysis of the trial samples collected, the authors concluded that the effect was due to an off- target rise in the concentration of an aldosterone-like factor, which resulted in an unanticipated increase in blood pressure. However, this is not entirely consistent with the fact that CHD mortality was inversely related to a raised blood pressure and the incidence of stroke was not greater in the treatment group. A second member of the CETP inhibitors, dalcetrapib, presented an opportunity to test the HDL hypothesis as this compound raised plasma HDL-C by 30–40 % but had little or no effect on LDL-C. A Phase III randomised controlled trial (Dalcetrapib: Dal-OUTCOMES; NCT00658515), involving 15,600 patients with recent acute coronary syndrome, began in 2008 but was halted in 2012, because of a perceived lack of efficacy55. Anacetrapib is the most potent CETP inhibitor to date and in the first clinical trial (Anacetrapib: DEFINE; NCT00685776)56 was shown to lower LDL-C by approximately 50 % and increase HDL-C by 140 %. Although, this gross effect may reflect an extreme disturbance of HDL-C metabolism and its consequences,the DEFINE trial did not report any significant increase in CVD in the test arm. However, this study was too small to provide robust information on the clinical events56.
Conclusion
Although two members of the CETP inhibitor drug family have clearly been removed from the drugs cabinet, it is still a fact that we do not know if the remaining members will provide a clinical benefit for patients with CVD/CHD. The two members of this family under clinical investigation, currently in Phase III randomised control trials (Anacetrapib: REVEAL; NCT01252953 and Evacetrapib: ACCELERATE; NCT01687998), are due to report in 2016/17. Results from these highly powered, multi-centre studies, will determine the success or failure of the original hypothesis that CETP inhibition is of benefit to CVD/CHD. Any chance of addressing whether success is through raising HDL-C per se is lost following the failure of Dalcetrapib, the only member of the family that had little effect of plasma LDL-C. Efficacy of either CETP inhibitor remaining in the arena, may be due to the ability of one or other of these drugs to further reduce plasma LDL-C, rather than a benefit of raising HDL-C concentration. Should the drugs fail to give a beneficial effect, the reasons will be largely unknown, but may relate to the altered kinetic of the dynamic effects that increase the circulating half-life and hence complexity of the HDL particles. The results of these studies are anticipated with gathering interest, and an additional benefit will be that they will provide an important opportunity to gain a further understanding of HDL metabolism/catabolism in disease, which will be invaluable in progressing with clinical development of these compounds.