





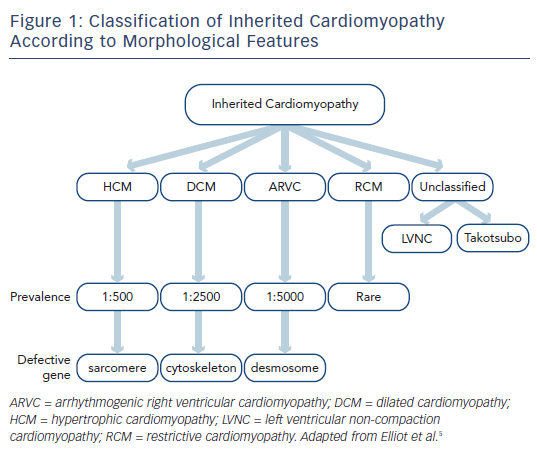
Inherited cardiomyopathies are primary disorders of the heart muscle that have a genetic basis. On long-term follow-up they are associated with adverse outcomes, particularly sudden death, arrhythmia and heart failure.1 The diagnosis is made on the basis of clinical, cardiac imaging (primarily with transthoracic echocardiography) and genetic features,2 ideally by a tertiary inherited cardiac conditions service that involves both cardiologists and clinical geneticists.3 Inherited cardiomyopathies are typically defined according to their imaging findings rather than their genetic basis4,5 and include hypertrophic, dilated, right ventricular, restrictive and unclassified phenotypes (see Figure 1).
The pathways that lead from genetic mutation to individual phenotypes of various cardiomyopathies have been studied for the past three decades. In broad terms, alterations within specific components of the cardiac architecture are associated with different cardiomyopathies, for example abnormal function of contractile proteins within the sarcomere is associated with hypertrophic cardiomyopathy (HCM), dysfunction of the link between the sarcomere and cytoskeleton is associated with dilated cardiomyopathy (DCM), and abnormal desmosome function is associated with arrhythmogenic right ventricular cardiomyopathy (ARVC).6 The expressed phenotype can vary dramatically between individuals, however, and mutations within the same gene can lead to different phenotypes. For example, different mutations in the beta-myosin heavy chain can lead to either HCM or DCM.7 Genetic testing should not therefore be used in isolation in the diagnosis of inherited cardiomyopathy and needs to be combined with clinical evaluation and cardiac imaging.
Due to its wide availability and safety, echocardiography forms the cornerstone of cardiac imaging in the diagnosis of inherited cardiomyopathy. With echocardiography it is possible to make an accurate assessment of cardiac morphology and function, which is necessary to make a diagnosis of an inherited cardiomyopathy. Echocardiography may be suboptimal in certain individuals, however, due to body habitus or hyperinflation of the lungs; in these individuals cardiovascular magnetic resonance (CMR) is recommended as an alternative.8 CMR is also recommended for assessment of the right ventricle (which can be difficult to assess in its entirety by echocardiography) and in certain cardiomyopathies such as left ventricular non-compaction and amyloidosis.8
A unique property of CMR is that it allows the interrogation of specific myocardial tissue characteristics, particularly the distribution of scar tissue. The presence of focal scarring is most widely assessed by the presence of late gadolinium enhancement (LGE). LGE imaging relies upon the administration of an exclusively extracellular gadoliniumbased contrast agent, which passively accumulates in damaged cells with a leaky cell membrane or areas of extracellular matrix expansion. The pattern of enhancement can aid in the diagnosis of the aetiology of cardiomyopathy as LGE has a characteristic appearance in many conditions.9 For example subendocardial or transmural LGE are seen in ischaemic cardiomyopathy, mid-wall LGE in DCM, patchy LGE in areas of hypertrophy in HCM, and global subendocardial LGE in amyloid cardiomyopathy (see Figure 2).
Extracellular fibrosis is a common pathological finding in many inherited cardiomyopathies. CMR to assess LGE is a well-established method by which to assess and quantify the extent of discrete areas of fibrosis. This process relies on the contrast between normal and abnormal myocardium, and a qualitative comparison of enhancement. When the myocardium is diffusely diseased, the LGE technique is limited and qualitative assessment can be insufficient to make a diagnosis. For such diffuse processes, quantitative tissue characterisation methods are available that measure the T1,10 T211 or T2* relaxation times of the myocardium.
Native tissue maps alone can be used for tissue characterisation. T1 mapping is often performed before and after the administration of a T1-shortening gadolinium-based contrast agent using techniques such as modified look-locker inversion recovery (MOLLI), shortened modified look-locker inversion recovery (ShMOLLI) and saturationrecovery single-shot acquisition (SASHA), which all have inherent strengths and weaknesses.12 Tissues with an expanded extracellular space due to fibrosis, infiltration or scarring have a larger distribution volume for the extracellular contrast agent, and consequently the reduction in T1 relaxation time is more pronounced than in normal tissue. Post-contrast myocardial T1 has been shown to significantly correlate with histological areas of fibrosis.13
From pre- and post-contrast T1 maps, the extracellular volume (ECV) fraction may be calculated as:

where ECV is the extracellular volume, Hct is haematocrit and R1=1/T1. ECV fraction has a significant correlation with the histological degree of fibrosis and volume of collagen in the myocardium.14
Native T1 mapping and ECV mapping both provide non-invasive assessments of tissue characteristics and can be used in conjunction with each other. ECV detects focal and diffuse extracellular fibrosis, however it requires intravenous cannulation for the administration of the contrast agent and measurement of haematocrit from a venous blood sample. Measurement of native T1 is not hampered by these issues, but in addition to extracellular fibrosis is also influenced by intracellular water, iron and fat, giving a weaker histological correlation with extracellular fibrosis than ECV.15 Furthermore, native T1 varies significantly depending on the field strengths, scanner vendor and technique used to measure it,16 and in clinical practice requires validation for the specific pulse sequence and field strength used.17
T1 Mapping in the Diagnosis of Cardiomyopathy
Extracellular matrix expansion and fibrosis are common pathological findings in many inherited cardiomyopathies.2 The non-invasive assessment of extracellular fibrosis by T1 mapping is therefore a potentially promising tool, in addition to conventional CMR including LGE, in the diagnosis of suspected cardiomyopathy.
Hypertrophic Cardiomyopathy
HCM is the most common inherited cardiomyopathy, affecting between two and 20 adults per 1,000.18 HCM is commonly defined as a disease of the cardiac muscle characterised by hypertrophy of the left ventricle in the absence of another cardiac or systemic cause.19 It is typically caused by autosomaldominant mutations of genes encoding contractile sarcomeric proteins and myofilament elements.20 These mutations lead to micro- and macroscopic changes within the heart, including cellular disarray, hypertrophy and interstitial fibrosis. These changes lead to hypertrophy that can affect the heart in a variety of patterns, and may affect any segment of the left ventricle.21 Although hypertrophy can be widespread, phenotypic expression within the same heart can be variable.22
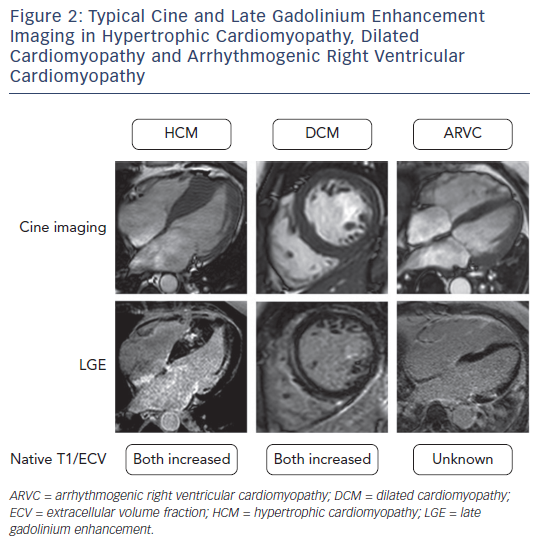
At present the diagnosis of HCM is primarily made by the identification of one or more hypertrophied segments by a cardiac imaging modality.18,19 In the majority of cases this involves identification of a ≥15 mm segment by echocardiography. CMR cine imaging enables accurate and reproducible assessment of the extent and location of hypertrophy when considering a diagnosis of HCM. It is reported that CMR detects hypertrophy in around 12 % of patients with HCM in whom it was not detected by echocardiography.22
LGE is seen in approximately two-thirds of patients with HCM and is typically found in areas of hypertrophy in a mid-wall pattern and at the right ventricular septal insertion points.23 The presence of LGE in HCM is suggestive of replacement fibrosis, and on necropsy there is a strong linear correlation between the extent of LGE within a particular segment and the amount of collagen measured histologically.24

With T1 and ECV mapping the extent of diffuse fibrosis in HCM can be quantified. Extracellular matrix expansion is one of the histological hallmarks of the disease process, and can be detected using quantitative T1 and ECV mapping techniques. Results demonstrate a good correlation with histological fibrosis quantification from specimens taken at the time of surgical myomectomy.14 One study has reported a correlation between post-contrast T1 time and histological degree of fibrosis in HCM hearts explanted at the time of transplant.25 However, in this study ECV (which is better validated than post-contrast T1 time) was not calculated, the sample size was small (four HCM necropsy specimens) and there was a long interval between the CMR and surgical explant.
Several studies have shown that both native T1 values26,27 and ECV14,28,29 are elevated in HCM. ECV is highest in regions with replacement fibrosis detected on LGE imaging. Native T1 value and ECV also appear to be elevated in regions without overt LGE,29,30 suggesting that they are able to detect more diffuse fibrosis not seen on LGE imaging. Several groups have also shown that both native T1 value and ECV are elevated in subjects with a mutation known to cause HCM without sufficient phenotypic expression to diagnose the disease on current imaging criteria.29,31,32
Differential Diagnosis of Hypertrophic Cardiomyopathy
Excluding alternative causes of hypertrophy of the left ventricle is fundamental to making a diagnosis of HCM and is often clinically challenging. Several of these “phenocopies” have a particular pattern on T1 mapping that could be clinically useful in differentiating them from HCM (see Figure 3).
Cardiac Amyloidosis
Cardiac amyloidosis is associated with progressive infiltration of the heart by extracellular amyloid protein resulting in hypertrophy of the left and right ventricles, interatrial septum and the atrioventricular valves. Given that this disease process involves pathological expansion of the extracellular space by amyloid protein, it is logical that both the native T1 value and ECV can be substantially raised in this condition.33–35 Patients with the highest native T1 value and ECV appear to have the greatest cardiac involvement and worst clinical outcomes.36 The typically very high native T1 value and ECV contributes to the differentiation of amyloidosis from HCM. In addition, the hypertrophy and fibrosis are more global in cardiac amyloidosis compared with the usually more regional hypertrophy in HCM.
Anderson-Fabry Disease
Anderson-Fabry disease is an inherited metabolic disorder associated with alpha-galactosidase deficiency and intracardiac glycolipid accumulation. The typical cardiac manifestation includes concentric biventricular hypertrophy, thickening of the atrioventricular valves and an inferolateral pattern of LGE. The disease process involves intramyocyte accumulation of lipids, which can shorten native T1 time. The extracellular matrix is not altered, however, and therefore recognition of a shortened native T1 value but normal ECV may aid in the diagnosis of Anderson-Fabry disease.37,38
Athlete’s Heart
Physical training leads to an increase in left ventricular mass and cavity size, and in certain situations this can be difficult to differentiate from HCM. HCM is the leading cause of sudden death in athletes and early identification is important so that abstinence from intense exercise can be advised.39 With T1 mapping it has been shown that as fitness increases in athletes there is an increase in myocyte mass with a relatively constant extracellular mass.40 As hypertrophy of the left ventricle increases in athletes there is therefore a decrease in ECV, whereas in HCM there is an increase in ECV. Based on this divergent principle, T1 mapping can be used to differentiate athletic remodelling of the left ventricle from HCM.41
Hypertension
Systemic hypertension can lead to concentric hypertrophy of the left ventricle and it is important that hypertensive heart disease is excluded before a diagnosis of HCM is made. It has been shown that in hypertensive patients regions of myocardial hypertrophy have increased native T1 values and ECV,42,43 albeit to a lesser extent than in HCM. These indices may therefore contribute to the differentiation of hypertensive heart disease from HCM.29 It should be noted, however, that in both HCM and hypertension as hypertrophy increases there is an associated increase in T1 indices, suggesting that the diagnostic utility may be lower in subjects with borderline hypertrophy. This is illustrated by the fact that both ECV and native T1 values are similar in subjects with hypertensive disease and in patients with a gene mutation known to cause HCM who do not yet have overt hypertrophy.29
Dilated Cardiomyopathy
DCM involves pathological dilatation and impairment of the left ventricle in the absence of abnormal loading conditions or ischaemic heart disease. A causative mutation can be found in around a third of patients with familial DCM.2 More than 40 causative genes, many of which relate to the cytoskeleton, have been identified.2 Mutation of these genes leads to loss of cardiomyocyte contractility, ultimately with the common pathophysiological end-point of cell death and fibrotic replacement.
The diagnosis of DCM is typically made after clinical examination and echocardiography. Fibrosis is a pathological feature of DCM and the CMR assessment of diffuse fibrosis by LGE and T1 mapping is increasingly being used in this situation. DCM is associated with a mid-wall pattern of fibrosis on LGE imaging, which when present confers an adverse prognosis.44 Mid-wall fibrosis is only present in around one-third of subjects with DCM, however, and T1 mapping may be a useful additional diagnostic tool in patients without overt changes on LGE.45 It has been demonstrated that both native T1 value and ECV are elevated in patients with DCM, even in regions without replacement fibrosis seen on LGE imaging.26,27,45 The native T1 value and ECV have been validated histologically in DCM against whole hearts explanted at the time of transplant15 and endomyocardial biopsy.46
One of the most common mutations in familial DCM is in the LMNA gene, which codes lamin A/C. In one study of seven families affected by a mutation in this gene it was shown that elevated ECV could be detected in gene carriers without overt left ventricular dysfunction.31
Part of the diagnosis of DCM involves the exclusion of alternative causes of cardiac dysfunction and dilatation, such as valvular or ischaemic heart disease. T1 mapping may have a role in the exclusion of other dilated phenotypes too, such as iron overload cardiomyopathy which has low native T1 value secondary to cardiac iron,47 and athletic ventricular dilation which has normal native T1 value and ECV.48
Elevated ECV has been shown to be an adverse prognostic marker in a mixed cohort of patients (including those with DCM but not HCM) and is associated with heart failure outcomes and mortality.49
Arrhythmogenic Right Ventricular Cardiomyopathy
ARVC is characterised by fibrofatty replacement of the right and sometimes left ventricles, which predisposes individuals to ventricular arrhythmia. Around half of ARVC cases are familial and the condition is typically caused by mutations affecting the desmosome.2 The diagnosis of ARVC is made by a combination of clinical, electrocardiographic, histological and imaging features.50 The diagnostic imaging features are regional right ventricular akinesia, dyskinesia or dyssynchrony in addition to dilation or impairment of function. In theory it might be possible to diagnose the fibrofatty replacement that is characteristic of ARVC as a shortened native T1 time; however the thin wall of the right ventricle poses important challenges and makes T1 mapping more susceptible to partial volume effects.
Other Inherited Cardiomyopathies
Familial restrictive cardiomyopathy is rare and is defined by a restrictive filling pattern on echocardiography. This phenotype may be caused by end-stage HCM or DCM, or can be caused by systemic diseases such as amyloidosis, sarcoidosis, carcinoid heart disease or scleroderma. T1 mapping could potentially be used as outlined previously to exclude alternative causes of left ventricular hypertrophy.
Left ventricular non-compaction cardiomyopathy (LVNC) is characterised by abnormal trabeculation, most commonly at the apex, and is often associated with ventricular hypertrophy, dilation or impairment of function. The molecular mechanisms of LVNC are not yet fully understood but genetic inheritance is reported in 30–50 % of patients.51 On CMR, a non-compacted to compacted myocardium ratio of >2.3 has been proposed as a diagnostic criterion for LVNC.52 As with ARVC, measuring T1 indices in the thinned compact layer of the myocardium is challenging and vulnerable to partial volume effects. One study has reported elevated native T1 values in a series of 31 patients with LVNC, even in areas without LGE.53
Takotsubo cardiomyopathy is characterised by acute transient apical ballooning of the left ventricle in the absence of obstructive coronary artery disease, often in the setting of psychological stress. The pathological processes are complex and likely involve catecholamine pathways, with some studies suggesting a genetic predisposition too.54,55 The transient myocardial dysfunction in Takotsubo cardiomyopathy appears to be mediated by myocardial oedema, which can be detected by a lengthened native T1 value in regions with oedema and dysfunction.56
Conclusion
T1 mapping is evolving as a useful clinical tool in the identification and diagnosis of inherited cardiomyopathy and it may have a future role in establishing the natural history of fibrosis progression in inherited cardiomyopathies and the timing of appropriate interventions. It is becoming particularly well established in the investigation of patients with unexplained left ventricular hypertrophy.
The optimal pulse sequence for T1 mapping is yet to be established, and standardisation remains an important aim. Standardised acquisition would also facilitate the publication of generally applicable normal ranges to facilitate more widespread use of T1 mapping in clinical practice and research. In the future, CMR sequences with shortened breath-holds or free breathing acquisition will be available to improve utility in patients with symptoms of dyspnoea. Sequences with improved spatial resolution will also aid in the investigation of T1 indices in thinned regions of myocardium, as in cases of ARVC and LVNC.
The prognostic importance of T1 indices over more commonly used imaging parameters such as wall thickness in HCM or ejection fraction in DCM remains to be established. Large prospective multicentre registries and trials are required to answer these questions and are awaited in the coming years.