





The sinoatrial or sinus node (SAN) is the heart’s natural pacemaker. Located in the superior right atrium, it automatically produces cyclical electrical activity to initiate each heartbeat in normal sinus rhythm. SAN dysfunction (SND) in humans, also known as ‘sick sinus syndrome’, can manifest as pathological bradycardia and asystolic pauses. As a result, SND can lead to symptoms of reduced cerebral perfusion such as dizziness and syncope. However, early SND may be latent and individuals may remain asymptomatic. Implantable electronic pacemakers are currently the only effective treatment. SND is the most common reason to have a pacemaker implanted, the indication for 27.5 % of all pacemakers implanted in the UK.1 The prevalence of SND in the UK is around 0.03 % affecting all ages, but it is much more common in the elderly population.2
The aetiology of SND can be intrinsic, extrinsic or often a mixture of the two. One retrospective study of 277 patients presenting to the emergency department with compromising bradycardia showed that 51 % of cases were attributable to a treatable extrinsic cause such as an adverse drug reaction, electrolyte imbalance or acute myocardial infarction. The other 49 % were assumed to be intrinsic or ‘idiopathic’.3
The pathophysiology of ‘idiopathic’ SND is still not clearly understood. Historically it is attributed to fibrosis and cell senescence and this is often still quoted today.4,5 However, contemporary evidence suggests that electrical remodelling of molecular pacemaking mechanisms such as membrane ion channels and intracellular Ca2+ cycling are important factors in SND.6 In this article we summarise the mechanisms of SAN function and review the current evidence surrounding the pathophysiology of SND.
Development of the Sinoatrial Node
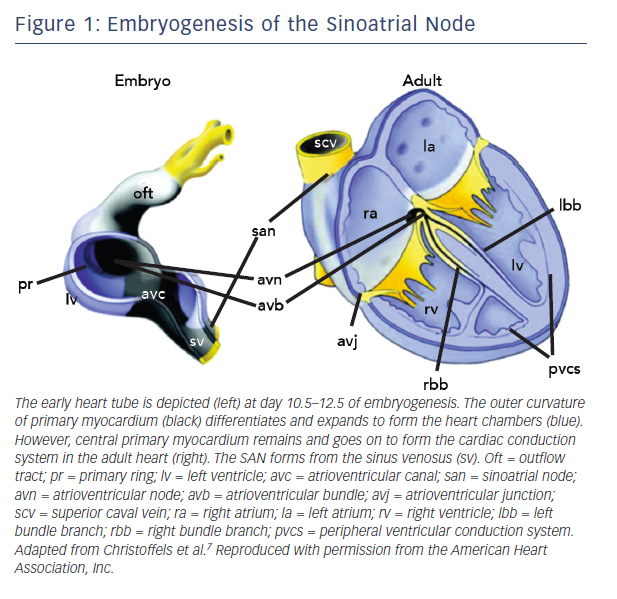
The SAN is the uppermost part of the cardiac conduction system (CCS), a chain of specialised tissue that directs electrical impulses through the heart and thus co-ordinates the way it contracts. The CCS is defined by a specific pattern of gene expression, differing to the surrounding ‘working myocardium’. During early embryogenesis as the heart tube forms, mesodermal cells quickly multiply and differentiate into working cardiomyocytes capable of contraction and fast conduction.7 However, the CCS is derived from primary myocardium that is instead led down a different lineage directed by specific transcription factors (Figure 1).7
Tbx3 is a T-box transcription factor found selectively within the CCS. Transgenic mice have been used to demonstrate its role in repressing working myocardial development and promoting a pacemaker programme of genes.8 These include key pacemaker genes, such as those encoding the low conductance gap junction connexin (Cx)45 and the hyperpolarisation-activated cyclic nucleotide-gated (HCN) membrane ion channel.8,9 The function of HCN channels within the SAN is discussed below.
The SAN is derived from an area of the developing CCS called the sinus venosus. The sinus venosus expresses a homeobox regulatory gene named Shox2.10 Shox2 represses the activation of Nkx2–5, Nppa and Cx40 which are all genes involved with contractile working myocardium.10 Nkx2–5 normally represses Tbx3 in the working myocardium and so Shox2 is associated with Tbx3 promotion.10 Null mutation of Shox2 in mouse embryos is lethal due to atrial malformation and severe bradycardia.10
Tbx18 is another important T-box transcription factor which appears during embryogenesis in the sinus horns of the sinus venosus and disappears from this region prior to birth.11 It drives progenitor cells in the sinus venosus to morphologically develop into a SAN core upon which Tbx3 then exerts its pacemaker programme of membrane ion channels.12 Tbx18-deficient mice have demonstrated a failure of this core SAN tissue to develop.12
Sinoatrial Node Structure
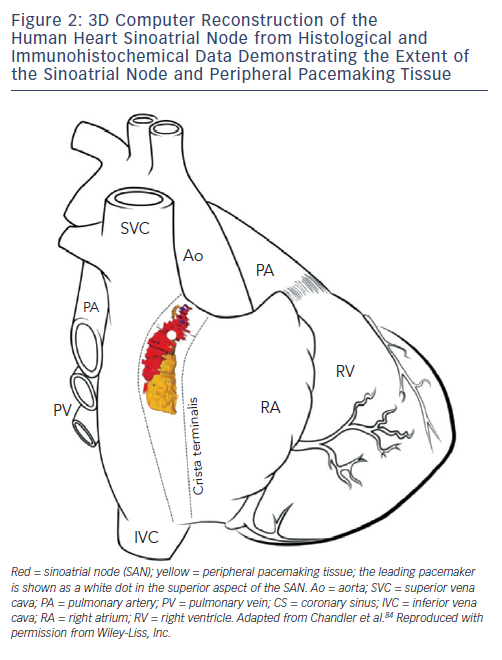
The SAN in humans is a much more extensive and complex structure than originally described (e.g. as portrayed in Figure 1). It is located 0.1–1 mm subepicardially within the posterior wall of the right atrium, closely opposed to the crista terminalis (CT), extending from near to the insertion of the superior vena cava (SVC) towards the inferior vena cava (IVC) (see Figure 2).13 The histologically defined human SAN ranges from 8–21.5 mm in length.13 However, nodal tissue capable of supporting pacemaker activity can be detected inferiorly as far as the most inferior aspect of the CT and the Eustacian ridge.14,15 The main body is crescent-shaped with a thinner tail of tissue extending below it.13 Nodal cells are densely packed within fibrous connective tissue.13 The cells are pale, small and relatively ‘empty’ of microfilament cytoskeleton and sarcomeric components compared with those of the surrounding muscular tissue.16
The SAN is a relatively small amount of tissue surrounded by a large amount of hyperpolarised atrial muscle. In order to deliver robust pacemaking it needs a way of effectively driving electrical activity into the right atrium and avoid being suppressed or becoming a source of re-entry. Transitional tissue within the SAN periphery forms a complex structure of block zones and exit pathways that allow co-ordinated delivery of stimulation to the right atrium. Recently optical mapping has demonstrated exit pathways at superior, middle and inferior levels from the SAN via which impulses are propagated into surrounding atrial muscle.17 This was correlated with histology showing insulating connective tissue and fat around the SAN except for where these exit pathways were exhibited functionally.17 Anatomically defined exit pathways are still controversial, however, since other work has failed to identify any histologically visible exit sites, suggesting a functional phenomenon.13,18
Tissue in the SAN periphery has several characteristics. Firstly, intermingling of nodal and atrial muscle cells has been seen with a gradual shift in the ratio of nodal to atrial cells (the mosaic effect).19 Interlocking ‘digits’ of SAN and working myocardial tissue have been seen.13 Additionally, the morphology of the cells themselves may gradually change between nodal and atrial cell types, with transitional cell types and intermediate features in between (the gradient effect).16,19 These features may help to gradually match SAN activity with the right atrium and promote an antegrade direction of conduction.20
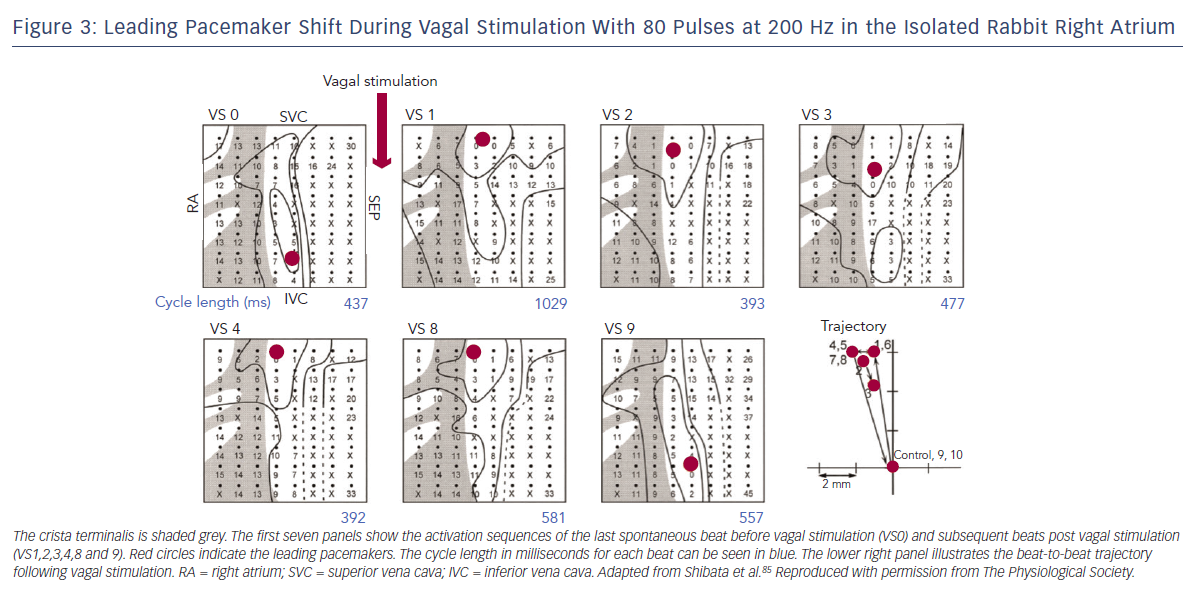
The first site of activation, termed the ‘leading pacemaker’, can shift within the SAN (see Figure 3). Although this site is usually in its superior aspect, it can sometimes be more inferior or even multifocal in some cases.21,22 Spontaneous pacemaking inferiorly in the SAN has been seen to be slower, and one theory is that there is a hierarchy of cells from those that fire fastest superiorly, to slower firing cells inferiorly.23 Heart rate changes can therefore be mediated by a shift in the leading pacemaker rather than by a single pacemaker site that changes its rate. For example, sympathetic stimulation can shift the leading pacemaker site superiorly within the SAN thereby increasing heart rate.23 This mechanism may also contribute to bradycardia in SND through a caudal shift in the leading pacemaker.24
Sinoatrial Node Automaticity
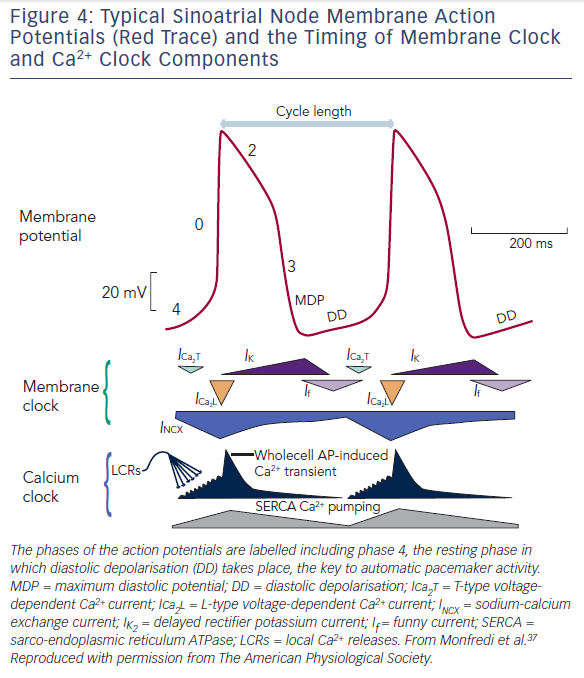
The SAN produces automatic electrical activity, which arises from several unique features. SAN cells have developed an interplaying combination of membrane ion channels, the ‘membrane clock’, and intracellular Ca2+ handling mechanisms, the ‘Ca2+ clock’, which lead to ‘diastolic depolarisation’ i.e. automatic depolarisation that occurs during the resting phase between beats (also termed ‘phase 4’ of an action potential). Phase 4 diastolic depolarisation serves to bring the membrane potential to the triggering threshold for the next beat and is key to SAN automaticity. Figure 4 shows phases 0 to 4 of the SAN action potential and summarises the timing of membrane and Ca2+ clock components as discussed below.
The Membrane Clock
Three membrane ion currents are key to the membrane clock – presence of the so-called funny current (If), decay of the delayed rectifier K+ current (IK,r), and absence of the inward rectifier K+ current (IK,1).
If is a mixed Na+ and K+ inward current that passes through membrane bound HCN channels. It activates early in phase 4 (diastole) when the cell is hyperpolarised and thus contributes to early diastolic depolarisation. Additional to this voltage-sensitive activity, HCN channels are also regulated by 3’-5’-cyclic adenosine monophosphate (cAMP) signalling and so are an important pathway for the regulation of heart rate by the autonomic nervous system.25,26 Hcn4 has been knocked out in transgenic mice by two groups with varied results. One group demonstrated ~50 % reduction in heart rate,27 while the other showed recurrent sinus pauses rather than profound bradycardia.28 Knocking out If does not abolish pacemaking completely, suggesting that although it is important for pacemaking, there are other mechanisms at play.
In order for If to depolarise the cell, opposing repolarising K+ currents (which normally bring working cardiomyocytes back to a stable resting potential) need to be absent or reduced. IK,1 is responsible for maintaining a stable resting membrane potential in working myocardium but it is absent in the SAN. Without IK,1, the SAN membrane potential is labile, which is key to aiding early diastolic depolarisation.29,30 Lastly, IK,r is responsible for repolarising working myocardial cells after the initial upstroke. This current is present in SAN cells and its decay aids early diastolic depolarisation.29,31,32
The Ca2+ Clock
Intracellular Ca2+ cycling is important in the excitation–contraction coupling of muscle cells, but in the SAN, Ca2+ contributes to pacemaking via the Ca2+ clock that is mutually entrained with the membrane clock, making an important contribution to automaticity.
Within SAN cells, the sarcoplasmic reticulum (SR) is central to the Ca2+ clock. Ryanodine receptors (RYRs) on the SR membrane release Ca2+ into the cell cytosol in the form of spontaneous ‘sparks’ or local Ca2+ releases (LCRs).33,34 These LCRs are seen to increase in frequency in response to Ca2+ influx through the cell membrane, termed Ca2+-induced Ca2+ release (CICR).33 Ca2+ influx occurs initially via T-type voltage-gated Ca2+ channels, which open as a result of the rising membrane potential initiated by the membrane clock.35 Thus LCRs lead to small increments in cytosolic Ca2+ concentration during phase 4. In response to rising intracellular Ca2+ levels, the Na+–Ca2+ exchanger (NCX) on the cell membrane exchanges one Ca2+ ion out for three Na+ ions into the cell leading to a net positive charge influx.36 In this way, the Ca2+ clock as a whole contributes to late diastolic depolarisation.
Finally, once diastolic depolarisation causes the cell membrane to reach threshold potential, phase 0 is triggered and L-type voltage-gated Ca2+ channels open allowing large scale depolarisation of the cell via Ca2+ influx.34 RYRs on the SR membrane respond again with CICR, emptying the SR en masse, leading to a whole-cell Ca2+ transient sweeping across the cell and myofilament contraction.34,37 Ca2+ stores in the SR are subsequently replenished by sarcoplasmic-endoplasmic reticulum calcium adenosine triphosphatase (SERCA) on the SR membrane.38
cAMP-driven pathways also play a major role in regulation of intracellular Ca2+ cycling, and so this is another way the autonomic nervous system can control heart rate.39 This among the other common links described between the membrane clock and the Ca2+ clock allow them to couple together to produce robust, automatic and cyclical diastolic depolarisation key to SAN automaticity.37
Diseases of the Sinoatrial Node
SND is normally diagnosed using the electrocardiographic features of bradycardia (<60 beats per minute) or asystolic pauses (>3 seconds) which may be due to reduced intrinsic SAN automaticity, sinus arrest or SAN exit block. Electrophysiology studies are not routinely performed in SND but may be done in equivocal cases. Two parameters, the corrected sinus node recovery time (cSNRT) and sinoatrial conduction time (SACT) in response to atrial pacing, can aid the investigation of SND. The sensitivity of both tests combined is 64 %, and the specificity if they are found to be significantly prolonged is 88 %.40 cSNRT is defined as the interval between the last paced beat and the next spontaneous beat, minus the spontaneous cycle length.41 A longer cSNRT indicates an increased tendency for suppression by ectopic activity from outside the SAN.
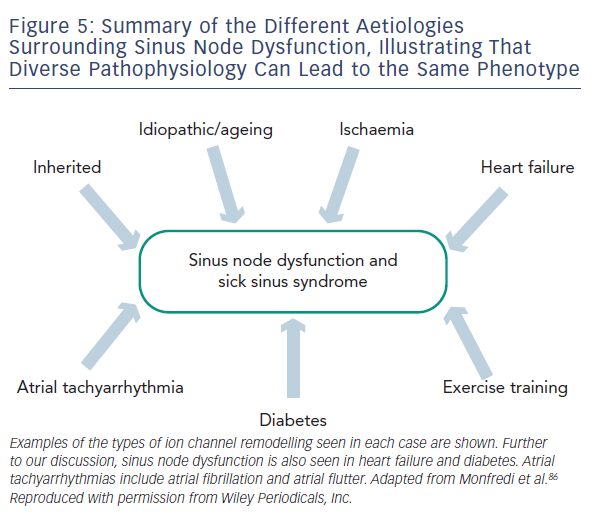
Regardless of the underlying pathology, SND is characterised by increased cSNRT, conduction delay along the CT, a more unicentric and caudal shift to leading pacemaker activity within the SAN and areas of low voltage within the right atrium attributed to atrophy and scar formation.42 In order to account for these changes in function, multiple processes and widespread remodelling are likely to be taking place, evidence for which is discussed below and summarised in Table 1 and Figure 5.
Idiopathic Sinoatrial Node Disfunction
Idiopathic SND predominantly occurs in the elderly population.43 SAN fibrosis is often quoted as the major cause. Histological studies in the 1970s of patients diagnosed with SND revealed that most cases were associated with either fibrosis or SAN atrophy.4,5 However, the same studies showed SND could also be associated with normal histology and, in some cases, severe fibrosis was associated with normal sinus rhythm.4,5 Furthermore, normal ageing of the SAN has been shown to be associated with SAN atrophy and fatty infiltration.44 It is therefore possible that at least some of the histological changes seen in SND cases were as a result of the ageing process itself. Clear causation of SND by fibrosis has not been established.
There is widespread electrical remodelling of the atria with ageing, and this has also been demonstrated in the SAN. This electrical remodelling is the result of changes in the expression of key ion channels that cause dysfunction of the normal pacemaker activity in the SAN. The Na+ channel Nav1.5 is normally present at the periphery of the SAN and is important for electrical coupling to right atrial myocardium.45 In ageing rats, Nav1.5 has been shown to decrease around the SAN periphery which could potentially lead to SAN exit block.46 Furthermore, the gap junction channel Cx43 is responsible for electrical coupling between working myocardial cells and is normally absent from the centre of the SAN. Ageing has been shown to reduce levels of Cx43 in the SAN periphery in association with conduction slowing.47
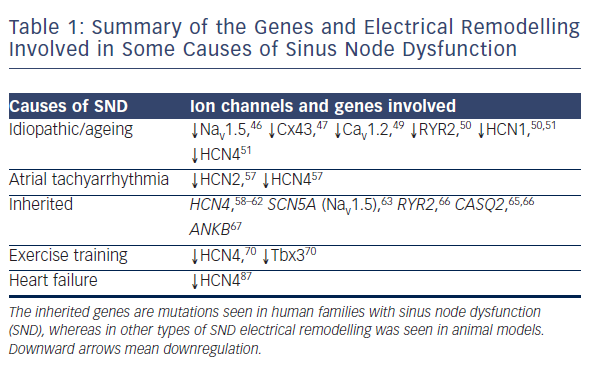
The L-type Ca2+ channel Cav1.2 is involved in the phase 0 upstroke of the SAN action potential.48 A comparison of young and old guinea-pigs demonstrated a decline in levels of this channel with ageing, progressing from the SAN centre outwards, and this was associated with increased sensitivity to the Ca2+ blocker nifedipine as well as reduced spontaneous SAN activity.49 Another study looking at young and old rats found decreased levels of RYR2 and the pacemaker channel HCN1, in association with a decreased intrinsic heart rate, increased action potential duration and cSNRT.50 Lastly, comparison of the functional and molecular features of young and old mice revealed bradycardia, increased SACT and reduced expression of a wide number of ion channels including HCN1, HCN4 and Nav1.5, along with several K+ and Ca2+ channels.51
Ageing is therefore associated with both structural and molecular remodelling and the cause of SND in this population is likely to be complex and heterogeneous.
Sinoatrial Node Dysfunction and Atrial Tachyarrhythmias
SND is often associated with intermittent episodes of atrial tachyarrhythmias such as atrial fibrillation (AF) leading to the term ‘tachy-brady syndrome’. Diffuse atrial remodelling can be seen in some patients with SND including atrial enlargement, increased refractoriness and prolonged conduction times which may predispose to the development of re-entrant circuits and AF.42 This suggests SND may be a prodrome of AF.
There is evidence that chronic AF causes SND by electrically remodelling the SAN. Patients undergoing electrical cardioversion for chronic AF demonstrate SND once reverted to sinus rhythm with bradycardia and increased cSNRT.52 In a canine model of atrial tachyarrhythmia using 20 Hz atrial pacing there is an increase in SNRT and a reduction of both maximal and intrinsic heart rate.53 The leading SAN pacemaker has been shown to shift caudally in patients with concomitant AF and SND, who also demonstrated a reduced sensitivity to sympathetic stimulation using isoprotenerol.54 The recurrence of AF after radiofrequency ablation has been predicted by the degree
of post-shock SNRT, which has been suggested as an indicator for the level of atrial remodelling.55 SAN function after cardioversion remodels to normal levels over time suggesting that SAN electrical remodelling in AF may be reversible in some cases.56 The molecular correlate of these changes is a reduction in mRNA expression of both HCN2 and HCN4 isoforms in the SAN. This was demonstrated in a canine tachycardia pacing model, with downregulation of these key pacemaker channels by >50 % in the SAN and correspondingly reduced If density by 48 % during patch clamp of isolated SAN cardiomyocytes.57
Familial Sinoatrial Node Dysfunction
Rare cases of inherited SND have allowed the analysis of specific gene mutations and subsequent channelopathies revealing their role in normal SAN pacemaking. DNA sequencing focusing on several candidate pacemaker genes has been used to screen patients with bradycardic phenotypes. These gene mutations give insight into their importance in SAN function for humans.
Several point mutations or deletions within the HCN4 gene have been found to be associated with bradycardia or paroxysmal AF.58–62 One study screened a patient with idiopathic SND, detecting a heterozygous single base deletion leading to a truncated C-terminus.58 When this mutant HCN4 gene was expressed in vitro in single cells, patch clamp experiments demonstrated If insensitive to increased cAMP levels.58 Three other studies focused on related patients each detecting three different mutations, all of which expressed mutant HCN4 channels in vitro that only activated at more negative voltages.59–61 Another study focused on patients with the same Moroccan-Jewish ethnic background, which revealed a novel point mutation that also led to HCN4 channels activating at more negative voltages.62 The studies looking at family or ethnic ties all suggested an autosomal-dominant inheritance pattern. Each of these mutations affected different aspects of the HCN channel and this demonstrates that these parts of the HCN channel are all required to function correctly for maintenance of a normal heart rate.
Mutations of the SCN5A gene lead to dysfunction of the Na+ channel Nav1.5, which is well known to be associated with a wide variety of cardiac diseases including long QT syndrome, Brugada syndrome, dilated cardiomyopathy, AF and SND.63 Since Nav1.5 is not specific to one area of the heart, some SCN5A mutations can cause a phenotype overlapping multiple syndromes in one patient.63 Although Nav1.5 is not present in the centre of the SAN, it is present in the periphery where it helps drive the action potential into the surrounding atrium, and so dysfunction of this channel may lead to delayed conduction, SAN exit block or sinus arrest.63,64 Multiple combinations of SCN5A mutations have been linked to SND with a suggested autosomal-recessive pattern of inheritance.63
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited condition, which leads to life-threatening stress-induced episodes of ventricular tachycardia causing syncope and sudden death in young patients without structural heart disease. There are at least two gene mutations thought to cause CPVT including genes for RYR2 and calsequestrin 2 (CASQ2) which are both involved with Ca2+ cycling in the cardiac SR. Patients with either of these mutations demonstrate normal electrocardiograms except for a significantly lower resting heart rate.65,66 As mentioned above SAN pacemaking is
known to depend on SR Ca2+ release and so bradycardia in CPVT is thought to be due to SR dysfunction within the SAN.65 Families demonstrating inherited RYR2 mutations show an autosomal-dominant mode of inheritance, whereas most cases of CASQ2 mutations show an autosomal-recessive pattern.65
Ankyrin-B (ANKB) mutations are associated with CPVT, long QT-syndrome and SND. One study reports on two families demonstrating long QT intervals, histories of sudden death, AF and pacemaker implantation due to severe bradycardia.67 Subsequent work in mice with heterozygous ANKB mutations demonstrated severe SND associated with either abnormal localisation or reduced expression of NCX1, Na+/K+-ATPase, inositol triphosphate receptor 3 (IP3) and Cav1.3.67
Athletes and Sinoatrial Node Dysfunction
Endurance athletes undergo a significant amount of exercise training and often exhibit profound sinus bradycardia as a result, with some even requiring pacemaker implantation later in life.68 This bradycardia is normally put down to high vagal tone i.e. is thought to be neurally mediated. However ‘intrinsic heart rate’, revealed by full autonomic pharmacological blockade, has also been shown to be lower in exercise trained individuals.69
A recent study demonstrated training-induced intrinsic bradycardia in rats and mice and showed a downregulation of Hcn4 and Tbx3 in these animals compared with controls.70 This demonstrates electrical remodelling as the mechanism for bradycardia in athletes and raises the question whether this could be a prodrome of SND. It is possible that these molecular changes are present in ex-athletes who require pacemaker implantation for SND later in life.
Ischaemia and Sinoatrial Node Dysfunction
Acute myocardial ischaemia involving the cardiac conduction pathways can commonly be seen to manifest in bradycardia due to abnormal autonomic tone, decreased perfusion or injury to SAN tissue.71,72 Ischaemia is often quoted as a cause of SND and disease of the main coronary vessels or sinus node artery has been implicated. Whether chronic ischaemia is a cause of SND is not clear. The incidence of both coronary artery disease and SND increase with age so patients may well have the two concomitantly. However, studies looking at the correlation in more detail show mixed results.
Post-mortem angiography has been used to compare sinus node artery patency in patients with SND and those without. It showed no significant obstructions in any patient, but reduced filling in about 20 % of patients with SND.73 One study comparing 46 patients with a history of inferior myocardial infarction showed that patients with severe sinus node artery stenosis (>75 %) demonstrated significantly lower intrinsic heart rates, longer cSNRTs and prolonged SACTs compared with moderate to no stenosis (<75 %).74 Another study demonstrated a lack of sinus node artery disease in patients with clinical SND yet severe sinus node artery disease in patients with normal SAN function.75 Furthermore, a post mortem study found no evidence of sinus node artery disease in any of its eight study patients with previously diagnosed SND.5 It is important to note therefore that a causal association between chronic ischaemia and SND has not yet firmly been established.
Future Prospects in Sinoatrial Node Research
Controversies remain in SAN research. Although we understand how membrane and Ca2+ clock pacemaker mechanisms are mutually entrained to produce diastolic depolarisation, there is still debate over the relative importance of each for maintaining pacemaker activity.76 Blockade or knockout of If does not abolish pacemaking whereas blockade of Ca2+ cycling does.28,36,77 On the other hand Ca2+ cycling mechanisms are not specific to pacemaker tissue whereas HCN channels can be used as a marker for the SAN.78 Further work is needed to clarify this contentious issue.
We have discussed the genes involved in SAN development including Shox2, Tbx18 and Tbx3, but there is still conflicting opinion about which genes have overarching control of this process. Tbx3 is thought to lead to expression of HCN channels in the CCS, but overexpressing Tbx3 in transgenic mice has a differential effect on inducing pacemaking depending on whether it is done in the embryo or adult, suggesting there are other mechanisms at play.79 Tbx18 is thought to primarily develop the morphology of the SAN head, but other groups have associated Tbx18 with HCN channel upregulation.7,80 These discrepancies suggest that our knowledge of the genetic control of pacemaking is not yet complete.
In the last 15 years or so the field of ‘biopacemaking’ has been progressing, striving to develop techniques that replicate pacemaking tissue ectopically via the manipulation of the molecular mechanisms discussed above. Initially individual genes important for pacemaking were introduced to non-pacemaking tissue, e.g. IK,1 knockout or HCN channel upregulation using adenoviral vectors.14, 81,82 However, pacing was often either too slow or unreliable for direct translation into humans. In an attempt to reproduce the complexity of the SAN, groups are now either modulating multiple features simultaneously or using transcription factors.83 Tbx18 and Tbx3 are prime candidates currently being studied in animals.8,11 Overexpression of Tbx18 has had particular success, reprogramming ventricular cells into SAN-like cells leading to robust and autonomically sensitive ectopic pacing in large animals.11,80 However, other than identifying the most effective gene, there are many challenges remaining before human trials can be successful. Questions remain about strategies for safe and stable gene expression, optimal location of delivery and how effective these strategies will be for each of the varying SND aetiologies.
Conclusion
Fibrosis, cell loss and coronary artery disease are often quoted as the main causes of SND. However it is clear that SND is not just one entity, and is rather the phenotype of many different disease processes. After over 100 years of studying the SAN and its disease we are still uncovering new insights into pacemaker function. Ion channel remodelling is now thought to be a major contributor to SND and the pattern of remodelling in different diseases can be wide and complex. A more complete understanding of the pathophysiology of SND will help us find ways to manipulate novel mechanisms in the search for alternative therapeutic options to the electronic pacemaker.