





Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy characterised by progressive replacement of the ventricular myocardium by fibrofatty tissue.1 Patients with the disease are predisposed to ventricular arrhythmias, heart failure and sudden cardiac death.
Pathophysiology
ARVC has a strong genetic basis with most disease variants displaying an autosomal dominant mode of transmission.2 Several mutations have been discovered to be implicated in familial variants of ARVC many of which encode proteins involved in cell–cell adhesion such as plakoglobin,3 desmoplakin4 and plakophilin-2.5 These findings have led to the understanding that this disease fundamentally arises from defects in the cardiomyocyte junction.6 The dysregulation of the cell junction is thought in itself to predispose patients to cardiac arrhythmias as well as result in cell detachment, apoptosis and subsequent replacement of normal myocardium with fibrofatty tissue particularly in the setting of mechanical stress. Fibrofatty replacement further interferes with electrical impulse conduction creating a substrate for ventricular arrhythmias.7
Clinical Presentation
Manifestations of ARVC vary widely with some patients being entirely asymptomatic while others experience debilitating arrhythmias, heart failure and sudden death. In patients with confirmed pathological mutations, the disease tends to manifest at an earlier age and follow a more aggressive trajectory.8
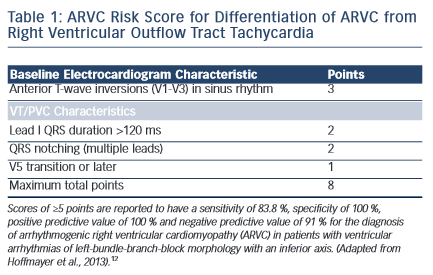
Common arrhythmias in patients with ARVC include frequent premature ventricular complexes (PVCs), non-sustained ventricular tachycardia (VT) and sustained VT.9,10 In those who develop VT, the majority of events are monomorphic and commonly originate in the right ventricle.11 In patients presenting with RV arrhythmias as the sole manifestation, it is important to differentiate between relatively benign aetiologies such as RV outflow tract tachycardia and ARVC as the treatment and prognostic implications are significant. Careful analysis of the electrocardiographic characteristics of the arrhythmia as well as the patient’s baseline electrocardiogram may provide important clues to the diagnosis. A recently published algorithm provides guidance in terms of this differentiation with excellent sensitivity and specificity (see Table 1).12
Symptoms experienced by ARVC patients include palpitations, dizziness and syncope as well as characteristic symptoms of heart failure in patients with advanced or long-standing disease. Mortality rates associated with ARVC have been reported to be as high as 2–4 % per year resulting from fatal arrhythmias and heart failure.13,14 Rates of sudden cardiac death are particularly high in ARVC with reports of up to 10 % of all sudden death cases in patients under 30 being attributed to this condition.15 The risk of sudden death appears to be especially high in patients who are young and may be the first symptom of the disease.16
Preventive Measures
Given the significant morbidity and mortality associated with ARVC, great effort has been expended to identify patients at particularly high risk for adverse outcomes and to develop therapies to improve their prognosis. In all patients with ARVC, primary intervention focuses on the prevention of disease progression. Patients are discouraged from participation in vigorous exercise as arrhythmias and sudden death events frequently occur at or around the time of exercise.17 Exercise additionally results in increased myocardial stress leading to the mechanical disruption of cell–cell junctions thus accelerating disease progression.18 For these reasons, many patients with ARVC are also prophylactically treated with beta-blockers, although no trial has demonstrated a significant mortality benefit for this therapy. Screening family members of patients with ARVC for clinical or genetic evidence of disease is highly encouraged as up to 50 % of relatives will test positive for the disease. Importantly, electrographic changes commonly precede structural changes, thus screening with an electrocardiogram may be effective in identifying early stages of the disease.19 In patients who develop symptoms, the mainstays of therapy have focused on antiarrhythmic medications, radiofrequency catheter ablation and the implantation of implantable cardioverter-defibrillators (ICDs).20
Risk Stratification and Therapeutic Options
Several retrospective studies have been conducted to identify high- risk features of the disease in order to guide therapy. Established high-risk features include significant RV dysfunction, left ventricular involvement, history of syncope and development of sustained VT.20–22 Of the available treatment modalities, only ICDs have consistently been demonstrated to affect patient mortality. In one study, the survival benefit of ICD implantation was close to 25 % over a 4-year follow- up period.23 A recent meta-analysis estimated the annual mortality rate of patients with ARVC who underwent ICD implantation at 0.9 %, substantially lower than those without ICDs.24 For this reason, patients with high-risk features are recommended to undergo ICD implantation by the American College of Cardiology, American Heart Association and the European Society of Cardiology.25 ICD therapy, however, does not decrease the rate of ventricular arrhythmias or disease progression. Additionally, ICD implantation carries a risk of procedural complications, tricuspid regurgitation and inappropriate therapy, which may contribute to patient morbidity. The annual rate of inappropriate therapy in those with ICDs has been estimated to be as high as 4 %.24 Therefore, the concomitant utilisation of catheter ablation and antiarrhythmic therapy is often necessary with the goals of reducing arrhythmia recurrence, decreasing ICD therapy and improving patient symptoms.
Catheter ablation has historically been effective in terminating malignant arrhythmias in the short term, but rates of VT recurrence following endocardial ablation are reported to be as high as 50–75 % within 3 years due to the progressive nature of the disease.26,27 Recognition of the larger role played by the epicardial arrhythmogenic substrate in ARVC has led to an increased focus on combined endocardial and epicardial ablation approaches. In a recent study comparing endocardial to endo+epicardial ablation, 83 % of patients treated with the combined approach remained arrhythmia free at 3 years compared with 52 % of patients treated with only endocardial ablation.27 Another study reported a similar success rate of 77 % with an endo+epicardial approach over an average follow-up of 18 months.28 While the results are very promising, it is important to recognise that epicardial ablation carries a substantial risk of complications such as epicardial bleeding and coronary stenosis occurring in approximately 5 % of cases.29 Nevertheless, catheter ablation remains an important therapeutic modality for decreasing patient morbidity in conjunction with ICD implantation and antiarrhythmic medication.
Antiarrhythmic Therapy
Individual Antiarrhythmic Therapy
Overall data on the use of antiarrhythmic agents in ARVC are relatively limited as no randomised clinical trials have been conducted to compare the efficacy of agents in this condition. Early studies investigating the use of antiarrhythmics in ARVC were small, focused on inhomogeneous patient cohorts with variable follow-up periods, and evaluated largely empirical medication choice.30–34
The first study to systematically assess the efficacy of antiarrhythmic therapy in ARVC was published in 1992 by Wichter et al.35 The initial study focused on 81 patients with proven or highly probable ARVC, but was later expanded to 191 patients in 2000.36 All patients underwent electrophysiological study and were tested for the inducibility of ventricular arrhythmias with programmed ventricular stimulation as well as the use of intravenous isoproterenol. Patients with both inducible arrhythmias and those without were then treated with a number of antiarrhythmic agents and reassessed for arrhythmia control. Drug efficacy was defined as increased difficulty of arrhythmia induction for patients with initially inducible arrhythmias and suppression of ventricular arrhythmias on 48-hour Holter monitor and exercise tests for patients with non-inducible arrhythmias. The patients were then started on antiarrhythmic therapy guided by these studies and followed for a number of months with assessment of arrhythmia recurrence and adverse events.
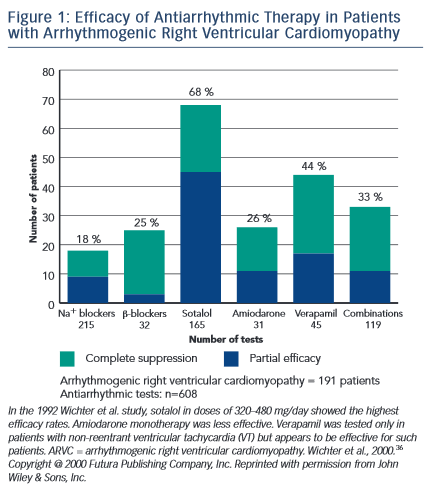
A total of 608 antiarrhythmic tests were conducted with various agents including beta-blockers, sodium channel blockers, verapamil, sotalol, amiodarone and combination therapy (see Figure 1). Sotalol, administered at a dosage of 320–640 mg/day, was determined to be the most effective therapy with approximately 68 % of patients achieving complete or partial arrhythmia suppression. Other therapies were less effective with Class I agents and amiodarone demonstrating only an 18 % and 26 % efficacy, respectively. Beta-blockers and verapamil proved to be most effective in patients with non-inducible arrhythmias on electrophysiology study and in patients thought to have triggered activity as the underlying mechanism of arrhythmia. In these patients, efficacy for these agents was 25 % and 44 %, respectively.
Based upon these observations, several conclusions were reached by the authors. First, sotalol appeared to be the most effective antiarrhythmic agent in the treatment of ARVC-associated arrhythmias. Second, amiodarone use should be limited given significant long-term toxicity and questionable efficacy. Lastly, patients with arrhythmias presumed to be brought on by triggered activity as opposed to re-entry may benefit from beta-blockers and verapamil.
These conclusions were further tested in a report from the North American ARVC Registry published in 2009 by Marcus et al.37 In this prospective cohort study, a group of 95 ARVC patients with implanted ICD devices were followed for 480 to 389 days. Patients were treated with a variety of antiarrhythmic medications selected at the discretion of their treating physicians. During the follow-up period, patients were contacted yearly for updates regarding changes in medications, symptoms, documented arrhythmias and ICD interrogations.
The major antiarrhythmic agents investigated in this study were beta- blockers, sotalol and amiodarone. A total of 58 participants received beta-blockers during the follow-up period; however, no significant difference was found in rates of clinically relevant arrhythmias compared with participants not receiving beta-blockers. Thirty-eight patients received sotalol but, contrary to Wichter et al.’s findings, these patients had no statistically significant difference in the rate of clinically relevant arrhythmias and even demonstrated a tendency towards increased arrhythmia rates. Only 10 patients were treated with amiodarone but experienced the most effective arrhythmia control with 75 % of patients benefiting from lower arrhythmia rates.
The disparate conclusions reached by Marcus et al. and Wichter et al. may be partially a result of significant differences in design of the two studies. First, the population in the Marcus et al. study may have been of higher risk given that all patients had definite ARVC with installed ICDs as opposed to Wichter et al.’s study in which none of the patients had ICDs. Second, the doses of sotalol used in the Wichter et al. study were on average much higher than in the Marcus et al. study (320–640 versus 160–320 mg/day). Likewise, the difference in the amiodarone results may have arisen from the fact that full amiodarone loading was not possible during the electrophysiology study period in the protocol utilised by Wichter et al. Third, the method of medication selection was less controlled in the Marcus et al. study relying on provider preference as opposed to the more standardised method of serial programmed stimulation utilised in the Wichter et al. study.
Despite the differences in their findings, these studies provide important guidance on the selection of antiarrhythmic therapy in ARVC. Both sotalol and amiodarone may represent effective antiarrhythmic choices in certain patients. Given the long-term toxicity associated with amiodarone its use should be limited in younger patients with a significant life expectancy; however, this risk should be balanced with the benefit of arrhythmia suppression. Beta-blockers and verapamil may be effective in patients with catecholamine-triggered arrhythmias although the efficacy of these agents in re-entrant arrhythmias appears to be limited.
Combination Antiarrhythmic Therapy
In patients who demonstrate poor response to individual agents, therapy with multiple antiarrhythmic medications may be considered. However, even fewer data exist to guide the selection of agents for use in combination therapy.
In the Wichter et al. study described above, a minority of patients were treated with combination therapy.35 In their cohort, the combination of Class I agents with amiodarone and sotalol were effective in a small number of patients in whom individual drug therapy had failed. Other reports indicate that the use of Class I agents combined with sotalol may be effective in controlling arrhythmias in those refractory to single agent therapy and failed endocardial ablation.38,39 One recent report demonstrated the effective addition of flecainide to patients receiving sotalol with resultant reduction in recurrent arrhythmias.38 Importantly, the addition of flecainide in this study was accomplished without significant adverse events despite a historic hesitation of using class Ic agents in patients with ventricular dysfunction stemming from experience with the post-myocardial infarction population. Since the publication of this report, an additional four patients in the authors’ cohort have been successfully treated with this combination and have likewise experienced excellent arrhythmia control without significant side-effects.
Several other studies have reported that the combination of amiodarone and beta-blockers may be effective in patients unable to achieve arrhythmia suppression with amiodarone alone. In a report by Tonet et al. following 31 patients with ventricular tachycardia, addition of beta-blocker therapy to amiodarone resulted in improved VT control in all patients.40 Only four patients in the study had documented ARVC, however. In another small study by Leclercq et al. focused on ARVC patients, the combination of amiodarone and beta blockers was likewise shown to result in VT suppression in all patients treated.39 It has been postulated that this combination is particularly effective due to the Class III and II action of the agents, which may work especially well in the catechomaline dependent arrhythmias in ARVC. This mechanism may also partially explain the efficacy of sotalol demonstrated in the Wichter et al. study.
Despite the promising results of several studies, much more research is necessary to establish the efficacy of combination therapy in treatment of ARVC. Additionally, toxicity of agents may increase when used in combination and thus such therapy should be used with caution. Nevertheless, in patients failing to achieve adequate arrhythmia control with an individual agent, combination therapy warrants consideration.
Conclusion
ARVC is a progressive disease that predisposes patients to ventricular arrhythmias, heart failure and sudden death. While no therapy exists to slow disease progression, treatment is aimed at reducing patient morbidity and mortality through the use of antiarrhythmic medications, catheter ablation and the implantation of ICDs. Only ICDs have been demonstrated to affect patient mortality, however antiarrhythmic medications are important in reducing patient arrhythmia burden and decreasing rates of inappropriate ICD therapy. Of the individual antiarrhythmics studied, sotalol and amiodarone appear to be the most effective in suppressing arrhythmias; however, the toxicities associated with long-term amiodarone use should be balanced against the benefit of arrhythmia reduction. Beta-blockers and calcium-channel blockers may be effective in catecholamine triggered arrhythmias but have limited efficacy in re-entrant rhythms. Monotherapy with Class I agents appears to be of likewise limited efficacy. In patients not achieving effective arrhythmia control with a single agent, combination antiarrhythmic therapy may result in improved outcomes. Particular combinations with demonstrated efficacy include Class I agents + amiodarone or sotalol as well as amiodarone + beta-blockers. Additional studies are necessary to provide further guidance regarding the use of antiarrhythmic agents in ARVC.